- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and discussion
Conclusion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2018; 2(4):403-416. doi:10.7150/ntno.28046 This issue Cite
Research Paper
MnO2-gated Nanoplatforms with Targeted Controlled Drug Release and Contrast-Enhanced MRI Properties: from 2D Cell Culture to 3D Biomimetic Hydrogels
Yupeng Shi, Flavien Guenneau, Xiaolin Wang#, Christophe Hélary, Thibaud Coradin ![]()
Sorbonne Université, CNRS, Collège de France, Laboratoire de Chimie de la Matière Condensée de Paris, 75005 Paris, France.
#Current address: State Key Laboratory of Quality Research in Chinese Medicine and School of Pharmacy, Macau University of Science and Technology, Macau.
Received 2018-6-24; Accepted 2018-9-8; Published 2018-9-21
Abstract

Multifunctional nanomaterials combining diagnosis and therapeutic properties have attracted a considerable attention in cancer research. Yet some important challenges are still to be faced, including an optimal coupling between these two types of properties that would be effective within complex biological tissues. To address these points, we have prepared novel nanoplatforms associating controlled drug delivery of doxorubicin and Magnetic Resonance Imaging (MRI) contrast-enhancement that exhibit high specificity towards cancer cells compared to normal cells and evaluated them both in 2D cultures and within 3D tissue-like biomimetic matrices.
Methods: Nanoplatforms were prepared from hollow silica nanoparticles coated with MnO2 nanosheets and conjugated with the AS1411 aptamer as a targeting agent. They were fully characterized from a chemical and structural point of view as well as for drug release and MRI signal enhancement. Standard two-dimensional monolayer cultures were performed using HeLa and Normal Human Dermal Fibroblasts (NHDF) cells to testify targeting and cytotoxicity. Cellularized type I collagen-based hydrogels were also used to study nanoparticles behavior in 3D biomimetic environments.
Results: The as-established nanoplatforms can enter HeLa cells, leading to the dissociation of the MnO2 nanosheets into Mn2+ that enhanced T1 magnetic resonance signals and concomitantly release doxorubicin, both effects being markedly more significant than in the presence of NHDFs. Moreover, particles functionality and specificity were preserved when the cells were immobilized within type I collagen-based fibrillar hydrogels.
Conclusion: The use of MnO2 nanosheets as glutathione-sensitive coatings of drug loaded nanoparticles together with surface conjugation with a targeting aptamer offers an effective strategy to obtain efficient and specific nanotheranostic systems for cancer research, both in 2D and 3D. The here-described tissue-like models should be easy to implement and could constitute an interesting intermediate validation step for newly-developed theranostic nanoparticles before in vivo evaluation.
Keywords: Silica nanoparticles, manganese dioxide, drug release, MR imaging, collagen hydrogels
Introduction
Despite the rapid improvement of modern medicine, the early diagnosis and therapy of cancer is still a challenge. The continuous development of nanotechnology and the emergence of targeted treatments provide an inequivalent opportunity in this area. The past decade has witnessed the engineering of many theranostic nanosystems, where the integration of different imaging agents and therapeutic drugs into a single nanoparticle (NP) has made it possible to exhibit multiple functionalities [1-4]. Extensive efforts have also allowed for the identification of active targeting reagents, such as folic acid, hyaluronic acid, aptamers, or transferrin, that bind with high specificity to the cancerous cell membrane [5-8]. At the same time, strategies were developed to achieve a controlled drug release triggered by intrinsic physiological microenvironment changes (pH, redox, enzyme, heat, etc.) and/or external stimuli (including light, magnetic/electric field, ultrasound, etc.) [9-11]. All those developments taken together, it becomes possible to simultaneously reduce the side-effects of anticancer agents to normal tissues and enhance their therapeutic efficiency [12]. Indeed, there still is some room for large improvement, in terms of drug loading and targeting efficiency.
From an imaging perspective, signals of most previously-reported nanosystems are “always on” regardless of the absence or presence of the target cells, resulting in a low contrast detection [13]. Among available nanoparticles, Mesoporous Silica Nanoparticles (MSNs) have been widely considered for the delivery of anticancer drugs [11,14,15]. To tackle MSNs intrinsic limited loading capacity, Hollow MSNs (HMSNs) with rattle or hollow structure have been prepared as they can efficiently accommodate drugs not only into mesoporous channels of their shell but also within their internal cavity [16-18]. Moreover, recent progress in the design of gated HMSNs has shown some promise in the development of controlled-release theranostic nanosystems. Different “gatekeepers”, such as organic molecules, biomacromolecules and nanoparticles, have been used, allowing pore-opening under the stimulation of pH change, temperature, nucleotides, antibodies, enzymes, glucose, or photoirradiation [19-21]. However, these gated nanosystems usually require a highly-sophisticated design that has a strong impact on synthesis time and development cost, ultimately hindering their transfer to the market, despite their demonstrated functionality.
Recently, MnO2 nanosheets, a 2D ultrathin semiconductor material with wide applications in the energy field [22], has attracted large attention as a gatekeeper for drug nanocarriers [23-25]. They exhibit a broad and intense absorption band at around 374 nm, making MnO2 nanosheets an efficient broad-spectrum quencher [26]. Moreover, MnO2 can be converted to Mn2+ via reduction by endogenous glutathione (GSH), leading to decomposition of the MnO2 nanosheets [27]. While manganese(IV) ions in the nanosheets are shielded from water due to their 6-fold coordination with oxygen atoms, free solvated Mn(II) ions are efficient T1 contrast agents for Magnetic Resonance Imaging (MRI) [28]. GSH is an essential endogenous antioxidant that has many cellular functions and high GSH levels are implicated in many diseases typically associated with cancer, liver damage, or heart problems [29]. Therefore, much attention has been paid to the use of GSH to design intracellular controlled release systems [30]. In addition, whereas the extracellular pH of normal tissues and blood is constant at 7.4, the measured extracellular values of most solid tumors range from pH 6.5 to 7.2, such lower pH values being more favorable for the reaction between MnO2 and GSH [31]. Based on this principle, several MnO2@SiO2-based controlled-release nanosystems with GSH-induced contrast-enhanced magnetic resonance signal have already been described [23, 32,33]. However, none of them took advantage of the specificity of HMSNs so far.
Another key challenge in this area lies in the precise control of the drug release using stimuli-responsive systems. Thus, tumor microenvironment, that has a crucial role in cancer progression, exhibit several specific biological and physico-chemical features that can be exploited to trigger the drug release at the tumor site [34]. However, the efficiency and stimulation of nanosystems are often tested in two-dimensional (2D) systems, in which seeded cancer cells do not experience the real microenvironment they find in vivo, where physiological fluids, tissues as well as interactions between cancer and stromal cells may impair the drug delivery or its functionality. The transfer from 2D data to in vivo during preclinical studies may therefore be extremely time-consuming and costly. Therefore, the design of three-dimensional (3D) environments exhibiting some features of in vivo tumors, such as three-dimensional architecture, cell-cell interaction and hypoxia should provide highly useful tumor tissue in vitro models for testing anticancer therapeutics [35,36]. In this context, the use of type I collagen, the major protein in most animal tissues, to prepare biomimetic constructs is of particular relevance [37-39]. Most importantly, it has been shown that they could act as models to study the interactions between nanoparticles and cells in a 3D environment [40,41].
Herein, we propose, for the first time, a novel and facile strategy for the fabrication of a multifunctional nanoprobe for contrast-enhanced bimodal cellular imaging and targeted therapy, as depicted in Figure 1. The nanoprobe consists of three components, including (1) doxorubicin(DOX)-loaded hollow mesoporous silica nanoparticles, (2) MnO2 nanosheets that act as both gatekeeper for DOX release from HMSNs and contrast agent for MRI and (3) cancer cell-targeting aptamers (AS1411), that bind to nucleolin, a nucleolar phosphoprotein which is overexpressed on the surface of certain cancer cells [28, 42, 43]. We demonstrate that this multifunctional HMSNs@MnO2(DOX)/apt theranostic nanosystem exhibits a synergistic delivery/imaging effect in standard 2D conditions. We also show that the functionality and specificity of these nanoplatforms are preserved within 3D cellularized collagen hydrogels, that can be useful biomimetic models for evaluating nanotheranostic systems performance before in vivo studies.
Schematic illustration of the preparation of HMSN@MnO2(DOX)/apt and the drug release mechanism.
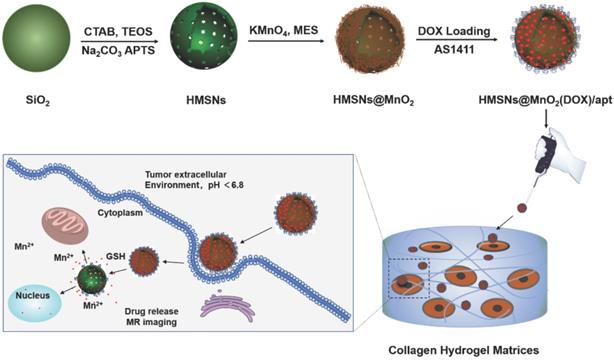
Methods
Materials and reagents
All chemical reagents were analytical grade and used without further purification. Cetyltrimethylammonium bromide (CTAB), tetraethyl orthosilicate (TEOS), sodium carbonate (Na2CO3), absolute ethanol, concentrated hydrochloric acid, ammonium aqueous solution (25-28%), triethanolamine (TEA), 3-Triethoxysilylpropylamine (APTES), potassium permanganate (KMnO4), 2-(N-Morpholino)ethanesulfonic acid hydrate (MES), doxorubicin hydrochloride (DOX) and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich. MilliQ water (18 MΩ, Millipore, France) was used for the preparation of the solutions and for all rinses. 4,6-diamidino-2-phenylindole (DAPI). Fetal bovine serum, Dulbecco's Modified Eagle's Medium (DMEM), trypsin, glutamine, penicillin-streptomycin solution and AS1411 aptamer (5′-NH2-GGTGGTGGTGGTTGTGGTGGTGGTGG-3′) were purchased from ThermoFisher.
Preparation of HMSNs@MnO2/apt
Bare HMSNs were prepared using previously reported methods [44,45]. To functionalize the particle surface with amine groups, as-synthesized HMSNs were first dispersed in 20 mL of toluene, followed by addition of 1 mL of APTES. The system was sealed and refluxed at 120 °C in oil bath for 12 h. Afterward, the mixture was centrifuged and washed with ethanol for several times to remove the residual APTES. Then, 20 mg of HMSNs-NH2 was dispersed in 4.2 mL MES buffer (0.1M, pH 6.0) and then 0.8 mL of 5 mM KMnO4 in water was added to the mixture under ultrasonic condition. The resulting mixture was sonicated for another one hour during which brown-black colloids were observed. Subsequently, the raw product was collected by centrifugation, washed several times with deionized water and alcohol to remove any possible residual reactants, and redispersed in 2 mL PBS solution (pH 7.4). The physical adsorption of aptamer on HMSNs@MnO2 was carried out by mixing 0.8 mL of HMSNs@MnO2 (20 mg/mL) in PBS and 200 µL of AS1411 (1 µM). After 3 h of incubation, the solution was centrifuged at 6000 rpm, washed several times, and then dispersed in PBS (pH 7.4) for further application. Full protocol is available in Supplementary Material. The preparation of fluorescently-labeled hollow mesoporous silica nanoparticles was performed by adding FITC to the starting solution used for bare HSMNs preparation.
Particles characterization
Transmission Electron Microscopy (TEM) studies were performed on a FEI Tecnai G2 Spirit microscope operating at 120 kV. Scanning electron microscopy (SEM) images were obtained on a Hitachi S-3400N scanning electron microscope with a field emission electron gun. Nitrogen sorption-desorption isotherms were measured at 77 K with a Micromeritics ASAP2010 analyzer. Fluorescence images were recorded on a LEICA microscope. Dynamic Light Scattering (DLS) was used to determine the hydrodynamic diameter of the nanoparticles in Milli-Q water or in culture medium with Mastersizer 3000 Particle Size Analyzer. The reading was carried out at an angle of 90° to the incident beam (632 nm). The Contin algorithm was used to analyze the autocorrelation functions. X-ray Photoelectron Spectroscopy (XPS) analyses were performed with a PHOIBOS 100 spectrometer from SPECS GmbH. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) was used to determine the Mn content of the nanoparticles.
Drug loading and release in vitro
10 mg of HMSNs@MnO2 nanoparticles were mixed with 1.5 mg of DOX in 1.5 mL of PBS/DMSO (1:1) mixture solution and then stirred under dark conditions for 24 h. Subsequently, the product was collected by centrifugation and washed several times with PBS to remove the free DOX. Then 100 µL of AS1411 (1 µM) in PBS was added, and after 3 h of stirring, the resulting HMSNs@MnO2(DOX)/apt nanoparticles were collected by centrifugation and re-dispersed in PBS solution for subsequent use. To evaluate the DOX loading, the remaining DOX molecules in the supernatant solutions were determined by fluorescence spectroscopy (λex= 500 nm, λem = 590 nm). The loading of the nanoparticles was expressed as the mass percentage of DOX with respect to the total mass of HMSNs@MnO2(DOX)/apt nanoparticles.
In vitro DOX release from the HMSNs@MnO2(DOX)/apt nanoparticles was studied in PBS buffer in absence or presence of GSH at pH values of 7.4 and 5.5. For each release study, 1.0 mL of DOX-loaded nanoparticles (1.0 mg.mL-1) were dispersed inside a dialysis bag that was soaked in 9.0 mL PBS and shaken at room temperature. At selected time intervals, the sample was collected and 2 mL of solution outside the dialysis bag was removed. Then, 2 mL of fresh PBS buffer was added. The removed solution was properly diluted and the amount of DOX molecules present was measured by fluorescence spectroscopy. The same amount of the DOX solution was introduced in the dialysis bag and its diffusion kinetics was used as control for drug release studies.
Cell culture
Normal Human Dermal Fibroblasts (NHDF) and HeLa cells were cultured in complete cell culture medium (DMEM with GlutaMAX™, without phenol red supplement, with 10% fetal serum, 100 U.mL-1 penicillin, 100 μg.mL-1 streptomycin). Tissue culture flasks (75 cm2) were kept at 37 °C in a 95% air: 5% CO2 atmosphere. Before confluence, the cells were removed from culture flasks by treatment with 0.1% trypsin and 0.02% EDTA. Cells were rinsed and resuspended in the above culture medium before use.
In vitro cytotoxicity assays
First, HeLa were seeded in a 24-well plate at a density of ~5×104 cells/well overnight to allow cell attachment onto the surface of the wells. Then, 0.5 mL of fresh medium containing various concentrations of HMSNs@MnO2(DOX)/apt was added into the wells. After incubation for 24 h, cell activity was evaluated by the Alamar Blue assay. Control experiments were performed by incubating HeLa cells with free DOX and HMSNs@MnO2(DOX) at various drug contents or HMSNs@MnO2(DOX)/apt at equivalent particle concentration for 24 h. To confirm the cytotoxicity of DOX released from the nanoplatform under GSH, similar control experiments were performed with NHDF cells incubated in presence of 5 mM GSH.
Cell uptake studies by fluorescence imaging
NHDFs and HeLa cells were seeded in a round glass disk that was inserted in 24-well plate (~5×104 cells per well) and cultured for 24 h. The cell medium was removed, and then cells were incubated with 0.5 mL of fresh cell medium containing FITC-labeled HMSNs@MnO2(DOX)/apt nanoparticles for another 12 h. After medium removal, the cells were washed with PBS for several times, and then 0.5 mL of fresh cell medium with or without 5 mM GSH was added and incubated for another 3 hours. Fixation of the cells was carried out using 4% (v/v) paraformaldehyde (PFA) for 2 h and rinsed three times using PBS. After washing, cellular nuclei were stained with 1% (v/v) solution of DAPI in PBS buffer for 20 min and washed with PBS three times. Last, cell imaging was carried out by fluorescence microscopy. For the HeLa cells imaging, the procedure was similar, except that HMSNs@MnO2(DOX) or HMSNs@MnO2(DOX)/apt particles were added and no GSH treatment was performed.
Measurement of MRI relaxation properties
Imaging of MRI phantoms measurement was performed with a Bruker Avance III HD 300 spectrometer, equipped with a 10 mm micro imaging probe, having a maximum gradient capacity of 3 T.m-1 in the x, y and z directions. The Multi Slice Multi Echo (MSME) pulse sequence was used, acquiring 1 echo but 8 slices with a slice thickness of 1 mm. The size of the images was: 128×128 with a Field of View of 9.5×9.5 mm, resulting in a voxel size of 74×74 µm. Images were acquired with 1 scan and T1 weighting was obtained by using a short repetition time (TR) of 200 ms and using the shortest possible echo time (TE), namely 4.92 ms, in order to minimize the effect of T2 relaxation.
Preparation of 3D collagen-based tumor models
Type I collagen was purified from rat tails and the final concentration was estimated by hydroxyproline titration, as previously described [37]. Tubes separately filled with collagen solution (2 mg.mL-1 in 17 mM acetic acid), whole cell culture medium, and 0.1 M NaOH were kept in ice bathes for 1 h before preparation to slow down the gelling kinetics of collagen. First, 500 μL of collagen solution and 400 μL of culture medium were added to a 1.5 mL tube and vortexed vigorously. After addition of 30 μL of 0.1 M NaOH and strong vortexing, 125 μL of the NHDF or HeLa cell suspension at a density of 106 cells/mL was added and mixed homogeneously. Then 0.9 mL was sampled from the mixture and deposited into a 24-well plate. The plate was then incubated at room temperature for 10 min for complete gelling of collagen.
Cytotoxicity and functionality within 3D models
Two sets of experiments were designed [41]. In a first approach, selected particles were introduced in the cell suspension prior to their addition to the collagen solution, at a final concentration of 100 μg.mL-1. Then collagen gels were formed by pH increase to 7. After 48 h of incubation, the cell viability was assessed in the same way as for 2D tests except that 800 μL of water were first added to the collagen gel, left for 0.5 h at room temperature in order to extract the Alamar Blue solution trapped in the gel, and then collected for the absorbance measurements. Particle internalization was also studied in the same configuration. For this, after the 48 h incubation period, the gels were rinsed 3 times with PBS and fixed with 4% paraformaldehyde overnight. Next, the fixed samples were dehydrated in ethanol and butanol, and incorporated in paraffin to be able to obtain 10 μm histological sections with a manual microtome. Before observation, the as-obtained samples were immersed in toluene, ethanol, and then water for rehydration. The cell nuclei were stained with DAPI for 10 min and rinsed with PBS before observation. The second approach consisted in adding 1.0 mL of a 0.2 mg.mL-1 suspension of the HMSNs@MnO2(DOX)/apt particles onto the surface of particle-free cellularized collagen gels. After 3 hours of contact, the MR imaging experiments were performed as described above.
Statistical Analysis
Graphical results are presented as mean ± SD (standard deviation). Statistical significance was assessed using one-way analysis of variance (ANOVA) followed by Tukey (compare all pairs of groups) or Dunnett (compare a control group with other groups) post-hoc test. The level of significance in all statistical analyses was set at a probability of P < 0.05.
Results and discussion
Preparation and characterization of the nanoplatforms
The HMSN@MnO2/apt nanoparticles were prepared according to the process depicted in Figure 1. In brief, monodisperse solid SiO2 nanoparticles (sSiO2) were first prepared using a modified Stöber method. The prepared sSiO2 were then coated with a CTAB/SiO2 shell via base-catalyzed hydrolysis of TEOS and condensation of silica onto the surface of CTAB pre-coated sSiO2. The resulting sSiO2@CTAB/SiO2 particles were simultaneously treated with Na2CO3 to remove sSiO2, and NH4NO3 to remove CTAB, resulting in HMSNs with hollow cores and penetrating pore channels [44]. APTES was grafted on the surface of nanoparticles to get HMSN-NH2. Finally, ultrathin MnO2 nanosheets were formed onto the surface of HMSN-NH2 thanks to the reaction with MES and KMnO4 to obtain HMSN@MnO2. DOX was then loaded inside or on the surface of the nanoparticles by the impregnation scheme described above. To end, AS1411 aptamers were attached on the surface of MnO2 nanosheets thanks to nucleobase-mediated physisorption [28].
As shown in the TEM image of Figure 2a, the prepared hollow mesoporous HMSN nanoparticles exhibit a uniform diameter of ~140 nm and form a colorless well-dispersed suspension in deionized water (DI) (Figure 2d). The diameter measured by DLS, Dm, is about 200 nm in DI, which is slightly larger than the TEM data and may reflect some aggregation (Table 1). The contrast between shell and core of the silica nanospheres confirms their hollow structure. The shell of the HMSN with a thickness of ~25 nm displays an obvious mesoporous silica structure generated by the removal of pore templates. In DI water, the obtained HMSNs exhibited a negative zeta potential ζ of -18.4 mV, as expected for silica surfaces (Table 1).
Mean diameter Dm from DLS with corresponding polydispersity index (PDI) and Zeta potential ζ in deionized water
| Sample | Dm DLS (nm) | PDI | ζ (mV) |
|---|---|---|---|
| HMSNs | 200 ± 18 | 0.254 | -18.4 ± 0.5 |
| HMSNs-NH2 | 256 ± 27 | 0.317 | +10.4 ± 0.3 |
| HMSNs@MnO2 | 319 ± 41 | 0.579 | -15.4 ± 0.2 |
| HMSNs@MnO2(DOX) | 316 ± 26 | 0.500 | -7.3 ± 0.8 |
| HMSNs@MnO2(DOX)/apt | 248 ± 15 | 0.279 | -16.1 ± 0.3 |
TEM images of (a) HMSNs, (b) HMSN@MnO2/apt, (c) HMSN@MnO2/apt after GSH treatment and the corresponding digital images from left to right (d, e, f).
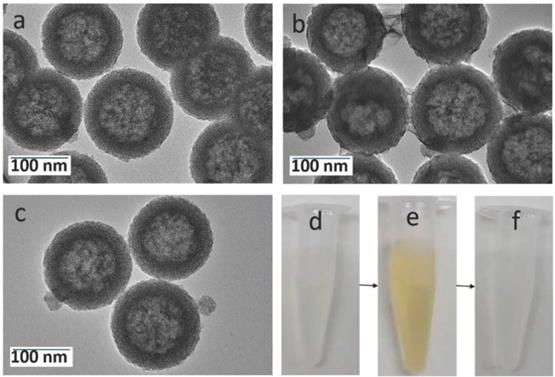
The N2 adsorption-desorption isotherm of the HMSN showed a typical type IV curve of surfactant-assisted mesoporous silica with a double strong and sharp adsorption step at intermediate relative partial pressure values around 0.4 (Figure S1). This feature is associated with nitrogen condensation inside the mesopores by capillarity. The application of the Brunauer-Emmett-Teller (BET) model resulted in a high value for the specific surface of 840 m2.g-1. In parallel, the Barrett-Joyner-Halenda (BJH) model applied on the adsorption branch of the isotherms led to an average pore diameter of 3.89 nm (Figure S1). The absence of a hysteresis loop in this interval of P/P0, as well as the narrow BJH pore size distribution, also suggested the existence of uniform cylindrical channels throughout the material.
The successful surface functionalization of the hollow mesoporous silica nanoparticles using APTES could be checked by the increase of the zeta potential that reached a positive value (+10.4 mV) (Table 1). Upon contact of these HMSN-NH2 particles with MES and KMnO4, a visual color change from white to brown was observed (Figure 2e). After extensive washing, UV-vis spectra clearly evidenced a strong absorption band at 400 nm, that can be attributed to the MnO2 nanosheets, whose intensity increase with the initial KMnO4 concentration (Figure S2). The formation of composite nanoparticles (HMSN@MnO2) with a rough capping layer was confirmed by TEM images (Figure 2b). Moreover, the average diameter of HMSN@MnO2 was changed from 200 nm to ca. 320 nm, accompanied by a polydispersity index (PDI) increase from 0.254 to 0.579, which suggests that the MnO2 coating induces a slight tendency for the particles to aggregate. The zeta potential of nanoparticles was also modified, from +10.4 mV down to -15.4 mV after functionalization, in agreement with values in the literature [46]. After coating, FTIR spectra also evidenced a peak at 1413 cm-1 characteristic of Mn-O vibrations in MnO2 (Figure S3). Sorption isotherms also confirmed the coating of the nanoparticles. HMSN@MnO2 presented flat sorption curves when compared to those of HMSN (at P / P0 = 0.4) (Figure S1), thus indicating a significant pore blocking. Nonetheless, HMSN@MnO2 showed an additional inflection at P/P0 > 0.8, that can correspond to secondary macropores resulting from the MnO2 capping layer with rough surface. HMSN@MnO2 showed a weak peak at pore diameter of 2.35 nm, confirming the partial pore blocking effect of the MnO2 capping layer. Finally, when HMSN@MnO2 were put in contact with GSH (10 mM), the color of the suspension changed back from brown to white, indicative of the disappearance of the MnO2 layer (Figure 2f). TEM images clearly confirmed the regeneration of the smooth surface and clear mesoporous structure of HMSN (Figure 2c).
DOX was then selected as a guest molecule to confer anticancer properties to the nanoparticles. The loading was performed by a simple impregnation route in a mixed aqueous/organic medium. The resulting DOX-loaded HMSN@MnO2 particles had a size distribution similar to unloaded particles but the zeta potential changed from -15.4 mV to -7.3 mV, suggesting that at least part of the drug is adsorbed on the MnO2 coating. The UV-vis spectra of the loaded particles showed an additional absorption band at ca. 480 nm similar to the one of DOX (Figure S2) and two peaks at 1624 cm-1 and 1580 cm-1 belonging to the benzene ring structure of DOX could be observed on the FTIR spectra of the HMSNs@MnO2(DOX) sample (Figure S3). The DOX loading of HMSN@MnO2, as estimated by fluorescent emission measurements of the supernatant after impregnation, reached ca. 80 µg.mg-1.
As a final step, the anti-nucleolin AS1411 was adsorbed on the HMSNs@MnO2(DOX) particles which resulted in a significant decrease in the average particle diameter and of the PDI (Table 1). This can be correlated with the more negative value of the zeta potential (from -7.6 mV without aptamer to -16.1 mV after AS1411 adsorption), that results in an enhanced particle colloidal stability thanks to electrostatic repulsion. FTIR spectra showed an additional peak at 1742 cm-1 that can correspond to the C=O stretching vibration of the aptamer (Figure S3).
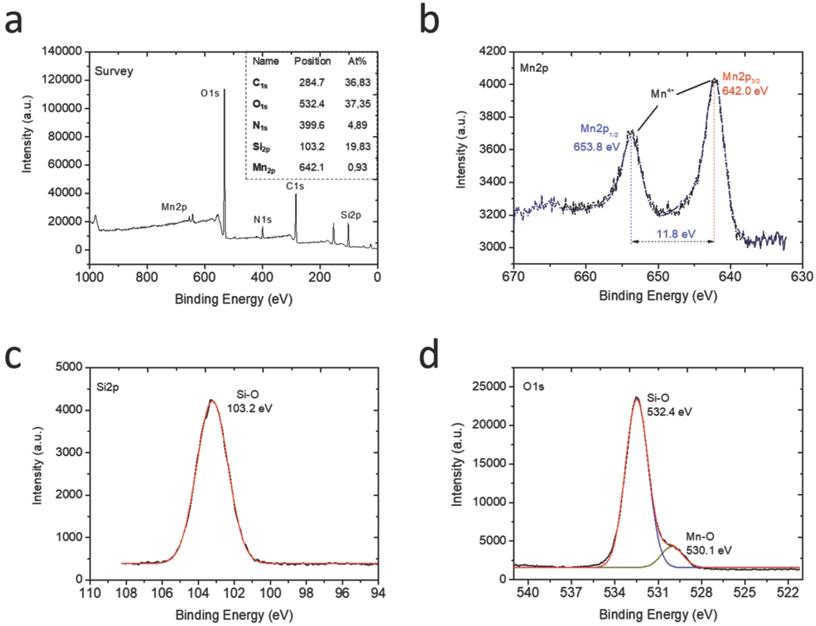
To characterize the chemical compositions of the final HMSNs@MnO2(DOX)/apt nanoparticles, their element composition was determined by XPS (Figure 3a). Mn, Si, C, O and N peaks were observed. Indeed, considering that the analysis depth of XPS analysis is 10 nm at maximum, it only probes the outer surface of the particles. It is therefore not surprising that the Si At% is rather low (19.8 %), corresponding to ca. 60 wt% of SiO2. In contrast, the contribution of organic molecules (DOX and AS1411) (> 35 % C) is enhanced compared to the whole particle volume. The Mn relative amount is 0.9 At% corresponding to ca. 5 wt % MnO2. Considering that manganese oxide nanosheets have been reported to be ca. 1 nm thick, a ca. 10 wt % amount would have been expected, suggesting that the MnO2 coating is not homogeneous over the whole particle surface. At the Mn2p level (Figure 3b), the two peaks at 642 eV and 653.8 eV unambiguously sign for the presence of MnO2, as also confirmed by the peak at 530.1 eV on the O1s spectrum [47] (Figure 3d). The peaks at 103.2 eV on the Si2p spectrum (Figure 3c) and 532.4 eV on the O1s spectrum are characteristic of hydrated silica [48]. Deconvoluted spectra at the C1s and N1s levels evidenced the presence of C-C, C-O, C=O, C-N and N-C=O groups (Figure S4). The latter is a clear indication of the presence of the aptamer on the particle surface [49], while the others can belong to both AS1411 and DOX.
XPS analysis of the HMSNs@MnO2(DOX)/apt nanoparticles: (a) full XPS spectrum with atomic analysis and deconvoluted signals with proposed attributions at the (b) Mn2p, (c) Si2p and (d) O1s levels.
Drug release and MRI imaging properties of the nanoplatforms
The feasibility of MnO2 degradation when treated with GSH and pH was first investigated. As shown in the Figure S5, the HMSNs@MnO2/apt nanoplatforms were stable at neutral pH but, after interaction with GSH, the color of the system quickly changed from brown to colorless, with a more pronounced effect when GSH concentration was increased from 5 mM to 10 mM. Acidic pH slightly destabilized the nanoplatform, due to the intrinsic instability of MnO2 in these conditions.
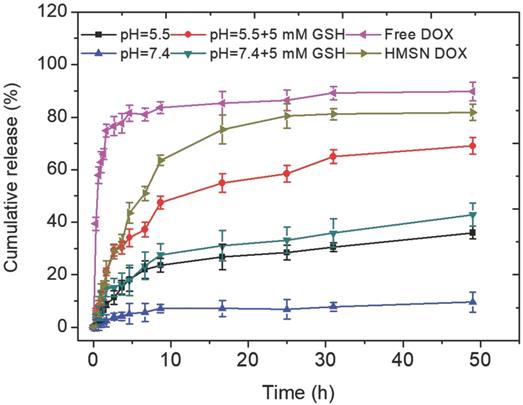
These observations were quantitatively assessed by monitoring DOX release in different conditions (Figure 4). In the absence of GSH, the DOX release was less than 10 % over 50 h at pH 7.4. When the MnO2 coating was destabilized, either in acidic conditions or by addition of GSH at neutral pH, ca. 20 % of the DOX content was released within 10 h and then a slower release, up to ca. 35-40 %, occurred in the next 40 h. When GSH was added in acidic conditions, the shape of the release curve was quite similar (i.e. fast release during 10 h followed by slower release) but the amount of released DOX at the end of the first phase was much higher (ca. 50 %) than in the two previous conditions and the total release after 50 h was larger than 60 %. As pointed out earlier, DOX molecules may be located on the MnO2 surface, inside the porous silica shell and within the particle empty core. Assuming that the GSH-free neutral pH conditions induce no significant dissolution of the MnO2 nanosheets, then the measured low release (less than 10 % of the initial dose) should correspond to surface-adsorbed DOX molecules. Upon GSH addition or acidification, much more DOX molecules are released following a two-step process. It is worth emphasizing that silica nanoparticles are less soluble in acidic than in neutral pH conditions and not sensitive to GSH unless specifically modified [50]. Thus it can be suggested that the first step corresponds to the release of DOX molecules both adsorbed on the surface and trapped in the porous shell due to the MnO2 coating dissolution, while the second one would sign for the slow diffusion of the drug located within the particles. However, when both acidic conditions and GSH addition are combined, the overall release profile, and especially the duration of the fast-release period, is not significantly modified but more DOX is initially released. This may be interpreted considering that, in these conditions, the MnO2 degradation is efficient enough to uncap all pores before diffusion of DOX molecules from the inside of the particle starts. In contrast, in GSH-only or acidic pH-only conditions, the MnO2 degradation process is less efficient and only part of the pores are uncapped before diffusion from the inside compartment becomes effective. This points out that the combination of reductive conditions due to GSH presence and acidic media where MnO2 is less stable is the most favorable for DOX release.
Cumulative release of the HMSNs@MnO2(DOX)/apt in different conditions: (blue) pH 7.4, (black) pH 5.5, (green) pH 7.4+ 5 mM GSH, (red) pH 5.5 + 5 mM GSH. Control samples: (pink) free DOX diffusion from dialysis bag and (pale green) uncoated HSMNs(DOX) in PBS solution (pH =7.4).
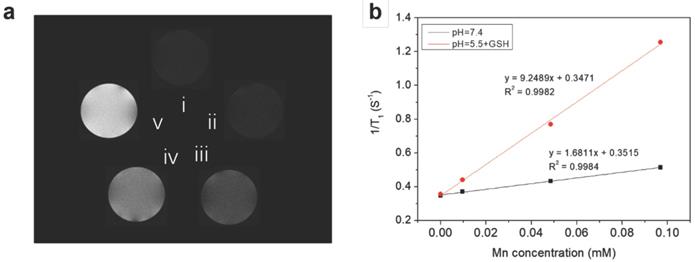
Because the reduction of MnO2 by intracellular GSH could produce a large amount of Mn2+, which are efficient T1 contrast agents, the nanoplatform could also afford GSH/pH-activated detection by MRI. To verify this hypothesis, T1-weighted MR images of HMSN@MnO2/apt aqueous suspensions under different conditions were first performed. As shown in Figure 5a, the MRI signal of the HMSN@MnO2/apt sample in PBS at pH 7.4 didn't exhibit an obvious T1-MRI signal difference compared to that of deionized water. A significant T1-MRI signal enhancement could be observed at pH 5.5. Adding 5 mM GSH in neutral and then acidic conditions further improved the T1-MRI signal intensity. The r1 relaxivity of the nanoplatform suspensions was also measured (Figure 5b) for HMSN@MnO2/apt at pH 5.5 in the presence of GSH, reaching a value of 9.25 mM-1.s-1 which is remarkably higher than that of the GSH-free solution at pH 7.4 (r1 = 1.68 mM-1.s-1). Importantly such r1 values are higher than those reported for other Mn-based nanocontrast agents (Mn3O4 nanoparticles, 8.26 mM-1.s-1; Mn-MSNs, 2.28 mM-1.s-1 and MnO nanoplates, 5.5 mM-1.s-1) [51-53]. The above results suggest that the MnO2 nanosheets can efficiently be converted into Mn2+ in a mildly acidic/GSH environment, explaining the rapid enhancement of the longitudinal relaxation rate with concentration. In conclusion, HMSNs@MnO2/apt present a clear pH/GSH-responsive T1-MRI performance.
(a) T1-weighted solution MR images of HMSN@MnO2/apt under different conditions. (i: H2O; ii: pH 7.4; iii: pH 5.5; iv: pH 7.4 + GSH 5 mM and v: pH 5.5 + GSH 5 mM) after incubation at 37 °C for 1 h. (b) 1/T1 versus Mn concentrations for HMSN@MnO2/apt at (black) pH 7.4 and (red) pH 5.5 + 5 mM GSH.
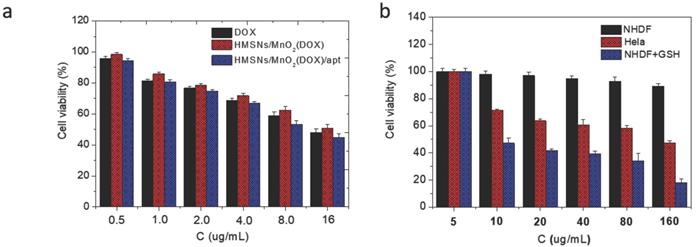
In a second step, it was necessary to check that the designed nanoplatforms could efficiently kill cancer cells while being safe for normal cells. First, it was checked that drug-free HMSNs@MnO2 nanoparticles had no impact on HeLa cells viability after 24 h of contact, as monitored by the Alamar Blue test (Figure S6). Then the cytotoxic effect of HMSNs@MnO2(DOX) and HMSNs@MnO2(DOX)/apt particles on HeLa cells viability was studied with the same method and compared to free DOX at the same drug concentrations. As shown on Figure 6a, no significant difference in cytotoxicity between free and encapsulated drug could be observed, with a continuous decrease of HeLa cells activity with increasing DOX dose, down to ca. 50 % for the highest investigated drug dose (16 µg.mL-1). The presence of the aptamer had no influence either. In a step further, the effect of HMSNs@MnO2(DOX)/apt particles concentration on HeLa and NHDF cells was monitored (Figure 6b). In the 5-160 µg.mL-1 range, HMSNs@MnO2(DOX)/apt particles have no obvious impact on NHDF while they are toxic for HeLa cells (i.e. viability below 80 %) for particle concentrations of 10 µg.mL-1 or more. Importantly, if GSH (5 mM) was present in the NHDF culture medium, then high cytotoxicity was also measured, confirming its ability to trigger DOX release.
(a) HeLa cell viability after incubation with DOX, HMSNs@MnO2(DOX) and HMSNs@MnO2(DOX)/apt for 24 h as a function of DOX dose. (b) Cell cytotoxicity of HMSNs@MnO2(DOX)/apt after 24 h of incubation with NHDF, HeLa and NHDF cells treated with GSH as a function of particle concentration.
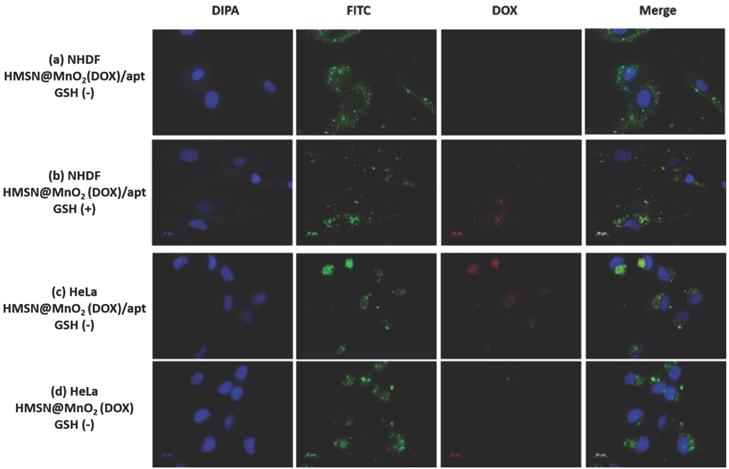
To confirm this hypothesis, the internalization of the HMSNs@MnO2(DOX)/apt was studied. As shown in Figure 7a-d, accumulation of the nanoplatforms, visualized by the green fluorescence of encapsulated FITC, in contact or within the cells could be observed for NHDF, without or with GSH, and HeLa cells. In the latter case, bright green aggregates are located near the nuclei, which appear blue after DAPI staining, strongly supporting their intracellular localization. For this sample, colocalization of the nanoplatform (green signal) and the DOX molecules (red signal) is evidenced. Noticeably, when no aptamer was present on the surface, low particle accumulation was observed and no clear red signal could be imaged (Figure 7d). For NHDF cells without GSH, the red areas are hardly seen whereas they are more clearly visible/detectable when GSH was present. It is important to point out that MnO2 nanosheets have previously been shown to efficiently quench the fluorescence of dyes in the UV-visible range [54]. Thus the observation of red signals on the fluorescence images indicates that DOX is no longer interacting with the manganese oxide coating, probably because the MnO2 nanosheets were reduced to Mn2+ by GSH. Noticeably, in the case of HMSNs@MnO2(DOX)/apt interacting with HeLa, the strong co-localization of FITC and DOX inside the cells would suggest that the MnO2 layer has been degraded but that part of the drug remains inside the particles (Figure 7c). In sharp contrast, for HMSNs@MnO2(DOX)/apt interacting with NHDF in the presence of GSH, many particles visualized by monitoring the green fluorescence do not exhibit a red fluorescence (Figure 7b). This can be attributed to the fact that the MnO2 layer is degraded by GSH leading to DOX release before the internalization process. While viability tests have evidenced NHDF cells death in these conditions (blue bars on Figure 6b), indicating that released drug could be uptaken, the dilution of DOX within the cytoplasm may lead to a low fluorescence intensity, precluding its detection.
Fluorescent images of (a) NHDF cells incubated with HMSNs@MnO2(DOX)/apt nanoparticles, (b) NHDF cells incubated with HMSNs@MnO2(DOX)/apt nanoparticles and 5 mM GSH, (c) HeLa cells incubated with HMSNs@MnO2(DOX)/apt nanoparticles, (d) HeLa cells incubated with HMSNs@MnO2(DOX) nanoparticles.
Altogether, HMSNs@MnO2(DOX)/apt nanoplatforms have the ability to be internalized by HeLa cells with a clear benefit of the presence of the targeting aptamer on the efficiency of this process. After degradation of the MnO2 coating, that may be due to both acidic pH of the lysosome and enhanced GSH expression, the particles can thereby release DOX molecules at the vicinity of the nucleus, resulting in an efficient cancer cell killing. At the same time, the formation of Mn2+ ions should allow for the detection of the cancer tissue via T1-weighted MRI. Very importantly, our experiments also indicate that these nanoplatforms are safe for normal NHDF cells in the absence of GSH.
Functionality of nanoplatforms in 3D biomimetic collagen matrices
At this stage of similar studies, in vivo evaluations are usually undertaken using tumor-bearing animals. However, in this work, we chose instead to go deeper into the understanding of the impact of a 3D environment on the functionality of the nanoplatform, a point that was not previously assessed in the literature. For this we prepared type I collagen matrices in conditions compatible with cell immobilization and particle encapsulation or diffusion [40,41].
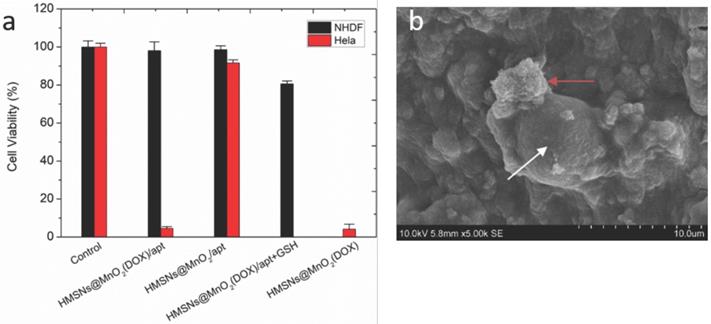
To check whether the nanoparticles preserved their efficiency and selectivity as cancer cell killing agents, the viability of NHDF and HeLa cells co-encapsulated with HMSNs@MnO2, HMSNs@MnO2(DOX), and HMSNs@MnO2(DOX)/apt at a dose of 100 μg.mL-1 within the collagen matrices was studied (Figure 8a). After 24 h, compared to the control group, the unloaded HMSNs@MnO2/apt showed very little toxicity for both types of cells. After DOX-loading, the HMSNs@MnO2(DOX)/apt nanoparticles neither showed much effect the viability of NHDF cells but exhibited a clear cytotoxic effect on HeLa cells, that was independent of the presence of the aptamer on the surface. Importantly, the decrease of HeLa cell viability was more significant than in the 2D experiments (see Figure 6b above), suggesting that the confinement of the cells and particles within the matrices strongly favors their interaction. As a matter of fact, SEM imaging allowed for the observation of nanoparticle aggregates at the close vicinity of the HeLa cells (Figure 8b). In such conditions, our results suggest that it may be no longer necessary to specifically target the cells by using the AS1411 aptamer.
(a) Viability of NHDF and HeLa cells within different types of 3D collagen/silica nanocomposites after 24 h of incubation. (b) SEM image of nanoparticles (red arrow) interacting with HeLa cells (white arrow) within collagen hydrogels.
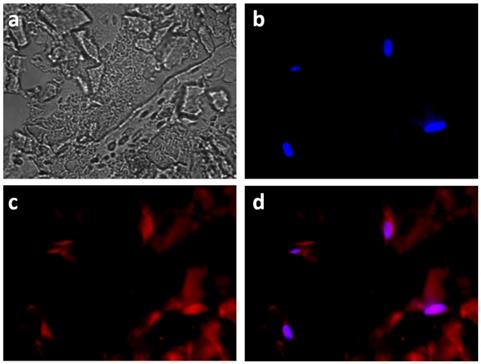
Fluorescence imaging of the encapsulated HeLa cells also evidenced the co-localization of DOX with the nuclei-staining DAPI, signing for their successful internalization (Figure 9). However red fluorescence signals could also be visualized outside the cells, suggesting that some MnO2 degradation also occurred outside the cells.
Fluorescent images of HeLa cells incubated with HMSNs@MnO2(DOX)/apt within a collagen hydrogel for 24 h, (a) bright field; (b) blue channel (DAPI); (c) red channel (DOX) and (d) merge.
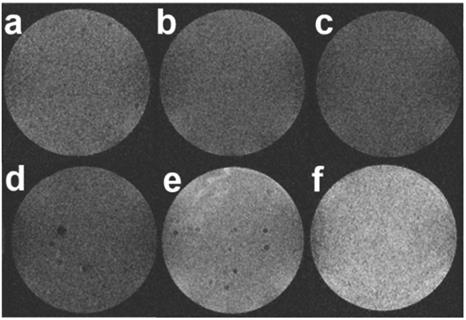
Then, to evaluate whether such an ultrasensitive GSH-responsive is suitable for in vivo tumor imaging by magnetic resonance, another configuration was used where selected nanoparticles were placed onto cellularized collagen hydrogels and left to diffuse for 3 h. As shown in Figure 10, a weak MRI signal was obtained for all samples prepared in the absence of HeLa cells or in the presence of NHDF cells, in agreement with the fluorescence image in Figure 9 that suggested partial destabilization of the nanoplatforms in contact with the collagen network. While comparable results were obtained when the aptamer-free particles were added to HeLa cells, HMSNs@MnO2(DOX)/apt particles led to a significant enhancement of the intensity of T1-weighted images, that was increased when the initial particle concentration was increased. These results clearly demonstrate that the functionality and specificity of the HMSNs@MnO2(DOX)/apt nanoplatforms is preserved within the 3D models. Interestingly, in this configuration, particles are not initially confined at the proximity of the cells but must find their way through the collagen network and bind to the HeLa cells. This explains why, in contrast to the previous system, the presence of the aptamer does have a significant beneficial effect on the MRI signal. In these 3D models, determination of the r1 value is difficult as it would require to determine the precise amount of HMSNs@MnO2(DOX)/apt that have diffused inside the matrices and interacted with the cells. However, when comparing Figure 5a and Figure 10, it quite clear that the T1 relaxation time is lower in the matrix than in suspension and that the decreased stability of the nanoplatforms in the collagen gels ultimately impacts on its contrasting efficacy, i.e. its ability to differentiate safe and tumor tissues.
T1-weighted MR images of collagen gels (a) without cells, (b,c) with NHDF cells and (d-f) with HeLa cells after 3h diffusion of (b,d) 0.2 mg.mL-1 HMSNs@MnO2(DOX) (c,e) 0.2 mg.mL-1 HMSNs@MnO2(DOX)/apt and (f) 0.4 mg.mL-1 HMSNs@MnO2(DOX)/apt.
Conclusion
As a conclusion, a multicomponent hybrid nanoplatform allowing for combined MRI contrast-enhanced imaging and targeted therapy through controlled drug delivery was designed, prepared and fully characterized in vitro in 2D and 3D configurations. Of particular importance is the fact that their high selectivity towards HeLa cancer cells compared to normal human cells was preserved when they were able to diffuse through cellularized biomimetic collagen hydrogels. This strongly suggests that such hydrogels, that are (i) simple to prepare, (ii) suitable for traditional cell biology studies, (iii) amenable to MRI measurements and (iv) can easily be implemented in terms of structure, composition and type of immobilized cells, should be useful for a preliminary screening of newly-developed nanotheranostics before entering in vivo studies that are time-consuming, costly and are raising increasing ethical and public concern.
Acknowledgements
Y.S. PhD grant was funded by the China Scholarship Council.
Supplementary Material
Full protocol for particle synthesis and supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064-79
2. Kelkar SS, Reineke TM. Theranostics: Combining Imaging and Therapy. Bioconjugate Chem. 2011;22:1879-1903
3. Muthu MS, Leong DT, Mei L, Feng S-S. Nanotheranostics ˗ Application and Further Development of Nanomedicine Strategies for Advanced Theranostics. Theranostics. 2014;4:660-77
4. Han L, Zhang X-Y, Wang Y-L, Li X, Yang X-H, Huang M. et al. Redox-responsive theranostic nanoplatforms based on inorganic nanomaterials. J Control Release. 2017;259:40-52
5. Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Deliv Rev. 2003;55:403-19
6. Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou S-F. et al. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine. Theranostics. 2015;5:23-42
7. Jiang T, Zhang Z, Zhang Y, Lv H, Zhou J, Li C. et al. Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials. 2012;33:9246-58
8. Dixit S, Novak T, Miller K, Zhu Y, Kenney ME, Broome A-M. Transferrin receptor-targeted theranostic gold nanoparticles for photosensitizer delivery in brain tumors. Nanoscale. 2015;7:1782-90
9. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nature Mater. 2013;12:991-1003
10. Cheng R, Meng F, Deng C, Klok H-A, Zhong Z. Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials. 2013;34:3647-57
11. Tarn D, Ashley CE, Xue M, Carnes EC, Zink JI, Brinker CJ. Mesoporous silica nanoparticle nanocarriers: biofunctionality and biocompatibility. Acc Chem Res. 2013;46:792-801
12. Fan Z, Fu FP, Yu H, Ray PC. Theranostic nanomedicine for cancer detection and treatment. J Food Drug Anal. 2014;22:3-17
13. Hu X, Liu G, Li Y, Wang X, Liu S. Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals. J Am Chem Soc. 2015;137:362-8
14. Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24:1504-34
15. Kim J, Kim HS, Lee N, Kim T, Kim H, Yu T. et al. Multifunctional uniform nanoparticles composed of a magnetite nanocrystal core and a mesoporous silica shell for magnetic resonance and fluorescence imaging and for drug delivery. Angew Chem Int Ed. 2008;47:8438-41
16. Chen Y, Chen H, Zeng D, Tian Y, Chen F, Feng J. et al. Core/shell structured hollow mesoporous nanocapsules: a potential platform for simultaneous cell imaging and anticancer drug delivery. ACS Nano. 2010;4:6001-13
17. Zhu Y, Shi J, Shen W, Dong X, Feng J, Ruan M. et al. Stimuli-Responsive Controlled Drug Release from a Hollow Mesoporous Silica Sphere/Polyelectrolyte Multilayer Core-Shell Structure. Angew Chem Int Ed. 2005;44:5083-7
18. Zhao W, Chen H, Li Y, Li L, Lang M, Shi J. Uniform rattle-type hollow magnetic mesoporous spheres as drug delivery carriers and their sustained-release property. Adv Funct Mater. 2008;18:2780-8
19. Cheng Y-J, Luo G-F, Zhu J-Y, Xu X-D, Zeng X, Cheng D-B. et al. Enzyme-induced and tumor-targeted drug delivery system based on multifunctional mesoporous silica nanoparticles. ACS Appl Mater Interfaces. 2015;7:9078-87
20. Zhang B, Luo Z, Liu J, Ding X, Li J, Cai K. Cytochrome c end-capped mesoporous silica nanoparticles as redox-responsive drug delivery vehicles for liver tumor-targeted triplex therapy in vitro and in vivo. J Control Release. 2014;192:192-201
21. Tang Y, Hu H, Zhang MG, Song J, Nie L, Wang S. et al. An aptamer-targeting photoresponsive drug delivery system using “off-on” graphene oxide wrapped mesoporous silica nanoparticles. Nanoscale. 2015;7:6304-10
22. Huang M, Li F, Dong F, Zhang YX, Zhang LL. MnO2-based nanostructures for high-performance supercapacitors. J Mater Chem A. 2015;3:21380-423
23. Chen Y, Chen H, Zhang S, Chen F, Sun S, He Q. et al. Structure-property relationships in manganese oxide - mesoporous silica nanoparticles used for T1-weighted MRI and simultaneous anti-cancer drug delivery. Biomaterials. 2012;33:2388-98
24. Zhang M, Xing L, Ke H, He YJ, Cui PF, Zhu Y. et al. MnO2-based Nanoplatform Serves as Drug Vehicle and MRI Contrast Agent for Cancer Theranostics. ACS Appl Mater Interfaces. 2017;9:11337-44
25. Jin L, Liu J, Tang Y, Cao L, Zhang T, Yuan Q. et al. MnO2-Functionalized Co-P Nanocomposite: A New Theranostic Agent for pH-Triggered T1/T2 Dual-Modality Magnetic Resonance Imaging-Guided Chemo-photothermal Synergistic Therapy. ACS Appl Mater Interfaces. 2017;9:41648-58
26. Zhang X-L, Zheng C, Guo S-S, Li J, Yang H-H, Chen G. Turn-on fluorescence sensor for intracellular imaging of glutathione using g-C3N4 nanosheet-MnO2 sandwich nanocomposite. Anal Chem. 2014;86:3426-34
27. Liu J, Meng L, Fei Z, Dyson PJ, Jing X, Liu X. MnO2 nanosheets as an artificial enzyme to mimic oxidase for rapid and sensitive detection of glutathione. Biosens Bioelectron. 2017;90:69-74
28. Zhao Z, Fan H, Zhou G, Bai H, Liang H, Wang R. et al. Activatable fluorescence/MRI bimodal platform for tumor cell imaging via MnO2 nanosheets-aptamer nanoprobe. J Am Chem Soc. 2014;136:11220-3
29. Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489-92
30. Meng F, Cheng R, Deng C, Zhong Z. Intracellular drug release nanosystems. Mater Today. 2012;15:436-42
31. Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56:1194-8
32. Yang X, He D, He X, Wang K, Zou Z, Li X. et al. Glutathione-Mediated Degradation of Surface-Capped MnO2 for Drug Release from Mesoporous Silica Nanoparticles to Cancer Cells. Part Part Syst Charact. 2015;32:205-12
33. Li X, Zhao W, Liu X, Chen K, Zhu S, Shi P. et al. MnO2-Based Mesoporous manganese silicate coated silica nanoparticles as multi-stimuli-responsive T1-MRI contrast agents and drug delivery carriers. Acta Biomater. 2016;30:378-87
34. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2-25
35. Edmondson R, Broglie JJ, Adcock AF, Yang L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol. 2014;12:207-18
36. Huang H, Zhang P, Chen H, Ji L, Chao H. Comparison between polypyridyl and cyclometalated ruthenium (II) complexes: anticancer activities against 2D and 3D cancer models. Chem Eur J. 2015;21:715-25
37. Hélary C, Ovtracht L, Coulomb B, Godeau G, Giraud-Guille MM. Dense fibrillar collagen matrices: a model to study myofibroblast behaviour during wound healing. Biomaterials. 2006;27:4443-52
38. Giraud-Guille MM, Mosser G, Hélary C, Eglin D. Bone matrix like assemblies of collagen: from liquid crystals to gel and biomimetic materials. Micron. 2005;36:602-8
39. Tidu A, Ghoubay-Benallaoua D, Lynch B, Haye B, Illoul C, Allain J-M. et al. Development of human corneal epithelium on organized fibrillated transparent collagen matrices synthesized at high concentration. Acta Biomater. 2015;22:50-8
40. Quignard S, Hélary C, Boissière M, Fullana J-M, Lagrée P-Y, Coradin T. Behaviour of silica nanoparticles in dermis-like cellularized collagen hydrogels. Biomater Sci. 2014;4:484-92
41. Wang X, Hélary C, Coradin T. Local and Sustained Gene Delivery in Silica-Collagen Nanocomposites. ACS Appl Mater Interfaces. 2015;7:2503-11
42. Bates P, Laber D, Miller D, Thomas S, Trent JO. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp Mol Pathol. 2009;86:151-64
43. Reyes-Reyes EM, Teng Y, Bates PJ. A new paradigm for aptamer therapeutic AS1411 action: uptake by macropinocytosis and its stimulation by a nucleolin-dependent mechanism. Cancer Res. 2010;70:8617-29
44. Fang X, Zhao X, Fang W, Chen C, Zheng N. Self-templating synthesis of hollow mesoporous silica and their applications in catalysis and drug delivery. Nanoscale. 2013;5:2205-18
45. Chen F, Hong H, Shi S, Goel S, Valdovinos H, Hernandez R. et al. Engineering of hollow mesoporous silica nanoparticles for remarkably enhanced tumor active targeting efficacy. Sci Rep. 2014;4:5080
46. He X, Yang X, Hai L, He D, He X, Wang K. et al. Single-layer MnO2 nanosheet quenched fluorescence ruthenium complexes for sensitive detection of ferrous iron. RSC Adv. 2016;6:79204-8
47. Militello MC, Gaarenstroom SW. Manganese Dioxide (MnO2) by XPS. Surf Sci Spectra. 2001;8:200-6
48. Soulé S, Bulteau A-L, Faucher S, Haye B, Aimé C, Allouche J. et al. Design and Cellular Fate of Bioinspired Au-Ag Nanoshells@Hybrid Silica Nanoparticles. Langmuir. 2016;32:10073-82
49. Petrovykh DY, Kimura-Suda H, Whitman LJ, Tarlov MF. Quantitative Analysis and Characterization of DNA Immobilized on Gold. J Am Chem Soc. 2003;125:5219-26
50. Quignard S, Masse S, Laurent G, Coradin T. Introduction of disulfide bridges within silica nanoparticles to control their intra-cellular degradation. Chem Commun. 2013;49:3410-2
51. Huang C-C, Khu N-H, Yeh C-S. The characteristics of sub 10 nm manganese oxide T1 contrast agents of different nanostructured morphologies. Biomaterials. 2010;31:4073-8
52. Peng Y-K, Tsang SCE, Chou P-T. Chemical design of nanoprobes for T1-weighted magnetic resonance imaging. Mater Today. 2016;19:336-48
53. Lee SH, Kim BH, Na HB, Hyeon T. Paramagnetic inorganic nanoparticles as T1 MRI contrast agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2014;6:196-209
54. He D, He X, Wang K, Yang X, Yang X, Zou Z, Li X. Redox-responsive degradable honeycomb manganese oxide nanostructures as effective nanocarriers for intracellular glutathione-triggered drug release. Chem. Commun. 2015;51:776-9
Author contact
![]() Corresponding author: Thibaud Coradin E-mail: thibaud.coradinfr; Tel: +33-1-44274018
Corresponding author: Thibaud Coradin E-mail: thibaud.coradinfr; Tel: +33-1-44274018