- Volume 10; 2026
- Volume 9; 2025
- Volume 8; 2024
- Volume 7; 2023
- Volume 6; 2022
- Archive
- Editorial Board
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusion
Experimental Section
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences

 Global reach, higher impact
Global reach, higher impact
Nanotheranostics 2019; 3(1):41-53. doi:10.7150/ntno.28450 This issue Cite
Research Paper
Intelligent Photosensitive Mesenchymal Stem Cells and Cell-Derived Microvesicles for Photothermal Therapy of Prostate Cancer
Liqun Huang1, Chang Xu2, Peng Xu3, Yu Qin4, Mengwei Chen2, Qishuai Feng2, Jing Pan4, Qian Cheng4, Feng Liang3, Xiaofei Wen1 ![]() , Ying Wang5, Yufang Shi5, Yu Cheng2
, Ying Wang5, Yufang Shi5, Yu Cheng2 ![]()
1. Department of Urology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, 200120, China.
2. Shanghai East Hospital; The Institute for Biomedical Engineering & Nano Science, Tongji University School of Medicine, Shanghai, 200120, China.
3. The State Key Laboratory of Refractories and Metallurgy, School of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Wuhan 430081, China.
4. Institute of Acoustics, Tongji University, Siping Road 1239, Shanghai 200092, China.
5. Key Laboratory of Stem Cell Biology, Institute of Health Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences/Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Received 2018-7-26; Accepted 2018-10-8; Published 2019-1-1
Abstract

Targeted delivery of nanomedicines into the tumor site and improving the intratumoral distribution remain challenging in cancer treatment. Here, we report an effective transportation system utilizing both of mesenchymal stem cells (MSCs) and their secreted microvesicles containing assembled gold nanostars (GNS) for targeted photothermal therapy of prostate cancer. The stem cells act as a cell carrier to actively load and assemble GNS into the lysosomes. Accumulation of GNS in the lysosomes facilitates the close interaction of nanoparticles, which could result in a 20 nm red-shift of surface plasmon resonance of GNS with a broad absorption in the near infrared region. Moreover, the MSCs can behave like an engineering factory to pack and release the GNS clusters into microvesicles. The secretion of GNS can be stimulated via light irradiation, providing an external trigger-assisted approach to encapsulate nanoparticles into cell derived microvesicles. In vivo studies demonstrate that GNS-loaded MSCs have an extensive intratumoral distribution, as monitored via photoacoustic imaging, and efficient antitumor effect under light exposure in a prostate-cancer subcutaneous model by intratumoral and intravenous injection. Our work presents a light-responsive transportation approach for GNS in combination of MSCs and their extracellular microvesicles and holds the promise as an effective strategy for targeted cancer therapy including prostate cancer.
Keywords: mesenchymal stem cells, microvesicles, gold nanostars, targeted transportation, photothermal therapy
Introduction
Prostate cancer is the most common non-skin malignant tumor among American men.1 As the age distribution in prostate cancer is mostly in elderly critical patients, a large number of them would suffer from a high risk of incontinence and erectile dysfunction after radical prostatectomy and radiotherapy.2-3 Although androgen depletion therapy could achieve moderate efficacy, not all of the patients are sensitive to the treatment and some of them would develop castrate-resistant prostate cancer.2-3 With the advance in imaging technology, such side effects can be reduced that leads to the progress of focal therapy in early-stage and low-grade prostate cancers.4 However, targeted and controllable treating the tumor foci with minimal side effects remains challenging in clinic.4 Therefore, a novel and efficient imaging guided antitumor therapy is urgently needed in the prostate cancer therapy.
Photothermal therapy (PTT), in which photosensitive agents are required to achieve selective thermal ablation in response of laser irradiation, has been widely investigated for cancer treatment.5-9 Among versatile photothermal agents, gold nanoparticles with tunable optical-electronic properties of surface plasmon resonance (SPR), efficient thermal energy conversion and good biocompatibility are promising candidates for PTT.10-11 Anisotropic gold nanostructures, such as nanoshells, nanorods, nanocages and nanostars have especially strong plasmon absorption in the near infrared (NIR) region to maximize the tissue penetration and offer a unique platform for photoacoustic imaging (PAI) and PTT.11-22 For example, gold nanoshell-mediated PTT is under clinical trials for focal ablation therapy of prostate tissues.12-13 Recently, researchers have shown that gold nanostars (GNS) with a 90% photothermal conversion efficiency and a surfactant-free wet-chemistry synthetic approach are promising photoabsorbing agents for PTT of cancers.18-19, 23-25 Effective targeting ability and broad tumor distribution of the nanoparticles are crucial to eradicate the cancer cells in PTT. Although these nanoparticles could accumulate in the tumor area due to the enhanced permeability and retention (EPR) effect, complex surface engineering approaches including physical adsorption and chemical conjugation are often required to improve the selectivity.17-18, 20-21 However, the accumulation in the tumor site remains a big challenge since most of the nanoparticles tend to be trapped by the mononuclear phagocyte system and removed from the blood stream.26-27
An alternative approach to enhance the accumulation and the distribution of nanoparticles at the tumor site is using tumor-tropic cells as biovehicles such as macrophages, platelets, neural stem cells (NSCs), induced pluripotent stem cells (iPSCs), and mesenchymal stem cells (MSCs) to overcome biological barriers.28-37 Among all kinds of cellular carriers, MSCs with multiple obtaining resources are widely utilized as an intelligent delivery platform for nanoparticles.35-37 Although researchers have shown the MSCs are able to transport the nanoparticles with good anti-tumor efficacy, the process of nanoparticle release from the carrier cells and uptake into cancer cells needs to be fully evaluated.35, 38 Furthermore, recent studies have discovered that the cell derivatives including microvesicles are natural biocarriers to protect and transport theranostic nanomaterials to the targeted cells.39-45 Several studies have investigated the function of microvesicles released from the nanoparticle-loaded endothelial cells and macrophages.39-44 These studies indicate that the secreted microvesicles might contain nanoparticles and could play a role in nanoparticle-loaded MSCs transportation.
In the present work, we hypothesize that the GNS-loaded MSCs could migrate and infiltrate the entire tumor site and release microvesicles containing GNS clusters for targeted transportation to cancer cells and PTT. In this work, we synthesized GNS with the TAT peptide on the surface for enhanced nanoparticle loading capacity (Scheme 1). These nanostars have excellent light-thermal conversion efficiency in the NIR region, biocompatible surface, and strong cellular penetration. MSCs loaded with the TAT-conjugated GNS (TAT-GNS) could facilitate the assembly of the nanostars in the lysosomes inside MSCs as an “engineering factory” and excrete the microvesicles loaded with GNS for tumor recognition and distribution.
Schematic design of MSCs-mediated delivery of GNS for photothermal ablation of cancer cells.
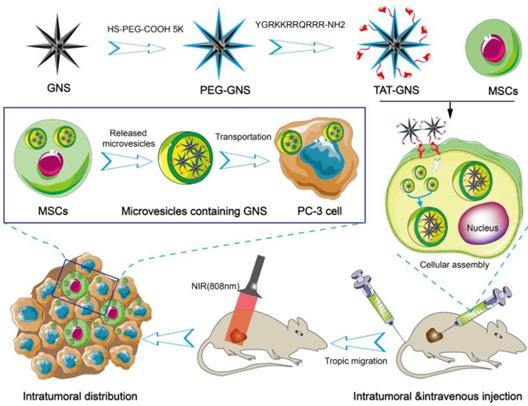
Results and Discussion
Synthesis and characterization of TAT-GNS
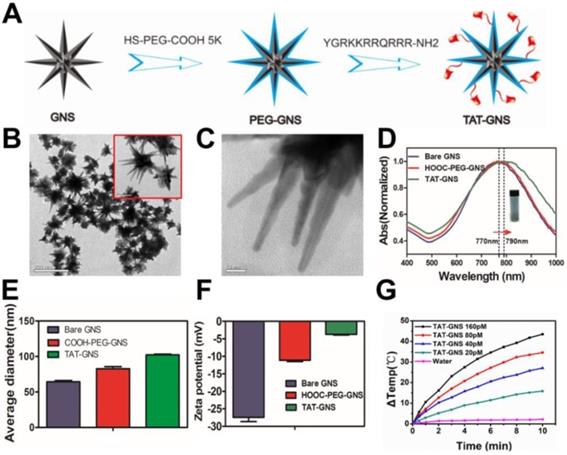
The GNS were synthesized through a seed-mediated growth approach and further modified with biofunctional HS-PEG-COOH (Fig. 1A).46 To increase cellular interaction, the trans-activating transcriptional activator (TAT) peptide, one of the most widely used cell penetrating peptides, was conjugated on the GNS surface via the amide bond formation. TAT-GNS was 82.5 ± 6.5 nm in diameter with sharp edges and tips as shown in Fig. 1B and Fig. 1C. This anisotropic structure provided a strong enhancement of plasmon absorption in the NIR region. As shown in Fig. 1D, the maximum absorption peak of TAT-GNS was around 790 nm, which showed a 20 nm red shift compared to the GNS and PEG-GNS. The GNS could be well dispersed in water (Fig. 1D). After functionalization, the GNS size also increased as measured by dynamic light scattering (DLS). Compared to the PEG-GNS with the average hydrodynamic diameter of 82.0 ± 1.7 nm, the size of TAT-GNS increased to 102.1 ± 0.6 nm (Fig. 1E). Successful TAT peptide conjugation was further confirmed via the zeta potential measurement. The ζ-potential of bare GNS was -27.47 ± 0.69 mV and PEG-GNS was -11.13 ± 0.22 mV. After the peptide modification, the surface charge of TAT-GNS became less negative with the ζ-potential of -3.74 ± 0.09 mV (Fig. 1F), suggesting that the TAT peptide with positive charges were conjugated on the surface of GNS. The effective functionalization was also verified by Fourier transform infrared (FT-IR) spectroscopy. TAT-GNS had the amide bond asymmetric stretching at 1577.1 cm-1 (Fig. S1). The time-dependent irradiation with the concentrations of TAT-GNS from 0 to 160 pM showed the excellent photothermal conversion under the radiation intensity of 2 W/cm2 (Fig. 1G). The TAT-GNS aqueous solution temperature could raise continuously upon irradiation and increasing of the nanoparticle concentration. After 10 min irradiation, the TAT-GNS solution at 160 pM had a 43.5 ℃ increase in temperature, which was sufficient to induce hyperthermia effect to cancer cells. The photothermal effect could also be adjusted via the changing of the laser irradiation intensities and the temperature change was positively correlated with the laser power (Fig. S2). In addition, the surface modification did not change the photothermal conversion efficiency of GNS as shown in Fig. S3. Both of PEG-GNS and TAT-GNS showed similar temperature raising curves.
Characterization of TAT-GNS. A. The scheme of TAT-GNS synthesis. B. The TEM image of GNS; C. The TEM image of the spikes of GNS. D. UV-vis-NIR absorption of TAT-GNS (The insert shows the TAT-GNS resuspension solution.); E. DLS size distribution of the bare GNS (grey column), HOOC-PEG-GNS (red column) and TAT-GNS (green column). F. Zeta potential of GNS, PEG-GNS and TAT-GNS; G. △T-t curve of TAT-GNS with different concentrations (rang from 0-160 pM) by laser irradiation with an intensity of 2 W/cm2.
TAT-GNS cellular uptake in MSCs
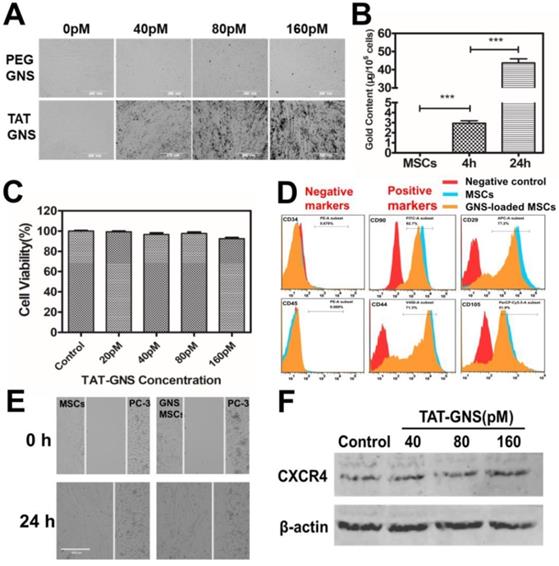
It is important to understand the interaction of TAT-GNS to the MSCs. Compared with the PEG-GNS, TAT-GNS showed enhanced intracellular uptake in MSCs visualized via the optical microscopy (Fig. 2A). The aggregates were observed at high TAT-GNS concentrations as shown in black dots in the optical images, indicating the cellular uptake of the nanoparticles. The cellular uptake of TAT-GNS was then quantified by ICP-MS. As shown in Fig. 2B, the gold content in the MSCs was 2.96 ± 0.41 μg/105 cells after 4 h incubation of 160 pM TAT-GNS. The nanoparticle uptake could be improved while prolonging the incubation time. After 24 h incubation, the gold content was 43.73 ± 3.86 μg/105 cells, which was 14.8 times higher than the 4 h incubation condition. Cytotoxicity of TAT-GNS to MSCs was evaluated by the CCK-8 assay (Fig. 2C). The MSCs maintained a high survival rate and over 92 % of cells were alive after incubation with TAT-GNS for 24 h. It indicates that TAT-GNS could have highly efficient uptake into the MSCs with little toxicity. The loading of nanoparticles into cells depend on their size, shape and surface chemistry.47-49 Various methods have been used to enhance the cellular loading efficiency of nanoparticles in stem cells, such as antibody modifycation, in-cell assembly and CPPs modification.33-35, 50-52 Here, TAT peptide, derived from the human immunodeficiency virus type 1 (HIV-1), was used for enhancing the interaction of GNS to stem cells. TAT peptide is widely used in enhancing cellular uptake of particles by actin-driven lipid-raft mediated micropinocytosis.46, 50, 53-55 Our results demonstrated that the TAT peptide modification could enhance the GNS loading capacity in the MSCs, in agreement with the expected effect of TAT peptide.
Cellular uptake and the tropic migration of TAT-GNS loaded MSCs. A. MSCs cellular uptake of PEG-GNS and TAT-GNS (ranging from 0-160 pM); B. ICP-MS of Au content in MSCs incubated with 160 pM TAT-GNS during 4 h and 24 h. C. Cell viability after labeling with TAT-GNS at different concentrations ranging from 20 to 160 pM. D. FACS analyses of the surface markers of labeled and unlabeled MSCs after 24 h incubation. E. Light microscopy images showing TAT-GNS loaded MSCs migration towards PC-3 cells. F. Western blot analysis of CXCR4 expression in GNS-labeled MSCs, β-actin as control.
Migratory ability of GNS-loaded MSCs
Tumor tropic migration ability is the key property of MSCs for targeted delivery. The cells loaded with TAT-GNS were analyzed by flow cytometry to identify the specific surface markers. MSCs were incubated with TAT-GNS for 24 h and collected for surface marker analysis. Positive staining markers for CD29, CD44, CD90, CD105 and negative staining markers for CD45 and CD34 were employed to analyze the stem cells (Fig. 2D).56 The results showed that the GNS-loaded MSCs did not show significant change in the expression of surface markers after loading the GNS in MSCs compared with MSCs. To further verify the stem cell function, the migratory ability of GNS-loaded MSCs toward prostate cancer cells was studied using the wound healing assay (Fig. 2E and Fig. S4). After 24 h observation, the gap between the GNS-loaded MSCs and PC-3 cells were merged, which was similar as the unloaded MSCs (Fig. 2E). The results showed that the migration of MSCs was maintained at the same level after loading of GNS. Consistently, the GNS-loaded MSCs could also migrate toward the other two prostate cancer cell lines DU 145 and LNCaP (Fig. S4). In addition, stromal-derived factor 1a (SDF-1a) and its receptor C-X-C chemokine receptor type 4 (CXCR4) plays an important role in the tumor tropism process and MSCs are positive for the expression of CXCR4.57 Therefore, the CXCR4 expression of GNS-loaded MSCs was measured after 24 h incubation with different concentration of TAT-GNS (ranging from 40 pM to 160 pM). No change was found in the expression of CXCR4 protein presented compared with that in the normal MSCs (Fig. 2F). All these results suggest that the viability and migration function of MSCs could be preserved after the GNS loading and utilized as the targeted delivery vector for treating prostate cancer.
TAT-GNS assembly in the MSCs lysosomes
The localization of TAT-GNS in MSCs was confirmed by confocal microscopy and TEM. To determine the location of the GNS in MSCs, FITC-TAT-GNS was prepared by utilizing FITC labeled TAT peptide instead of TAT peptide. After 24 h incubation, the TAT-GNS accumulated and aggregated in the MSCs. The GNS showed good colocalization with lysotracker red, indicating the nanoparticles mainly localized in lysosomes (Fig. 3A) and more images could be seen in the supplementary materials (Fig. S5). In addition, the TAT-GNS loaded MSC suspension displayed a maximum plasmon absorption at 810 nm. Compared with the TAT-GNS, TAT-GNS loaded MSCs showed a broader plasmon absorption and 40 nm red-shift of the maximum absorption in the NIR was observed (Fig. 3B). It suggested that the interparticle distance of GNS in the lysosomes was close enough to induce the SPR coupling, which guided us to explore the accumulation status of GNS inside the MSCs.
Cellular localization of TAT-GNS in MSCs and the photothermal property of TAT-GNS in MSC lysosomes. A. Laser scanning confocal microscopy images of the FITC-TAT-GNS loaded MSCs. B. UV-Vis-NIR spectrum of GNS-loaded MSCs (3×105 cells) suspension in PBS (1 mL). C. representative TEM images of MSCs incubated with TAT-GNS for 24 hours. The aggregation state of GNS in the lysosomes of MSCs. And the distance between the sharps of GNS inside the MCSs were determined by TEM. D. the △T-t curve of GNS-loaded MSCs (3×105 cells) suspension in PBS (1 mL) under the optical density of 2 W/cm2.
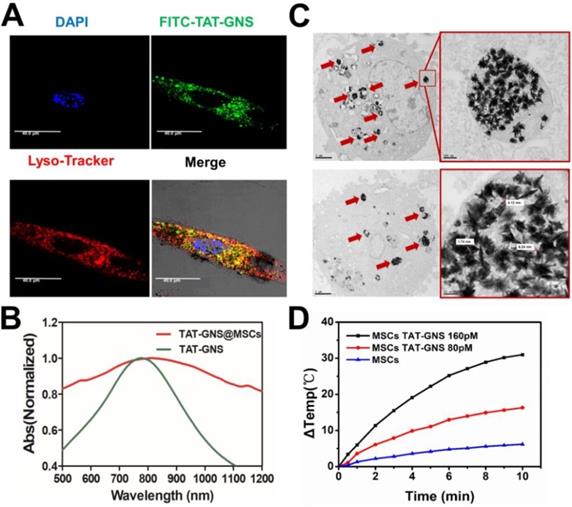
The intracellular distribution of TAT-GNS in the lysosomes was further studied by TEM. As shown in Fig. 3C and Fig. S6 (denoted by red arrows), TAT-GNS were located in the lysosomes after incubation, which was consistent with the confocal microscopy results. The GNS maintained their shape and were closely packed in the lysosomes. The distance between two nearby GNS ranged from 0 nm to 10 nm, which could induce strong plasmon coupling in the closely packed GNS clusters.58-59 Moreover, no GNS were observed in the cell nucleus and cytoplasm. After packaged in the lysosomes, the GNS also had an excellent photothermal conversion efficiency (Fig. 3D). The temperature of the cell suspension incubated with 160 pM TAT-GNS could raise almost 30 ℃ post 10 min irradiation at 2 W/cm2. It suggests that the cell internalization of GNS maintained their photothermal efficiency and could be utilized to damage cancer cells via hyperthermia effect. The mechanisms to intracellular assembly are likely attributed by the attraction of van der Waals forces and the compacting pressure in the lysosomes.
Excreted microvesicles of MSCs containing GNS clusters for cancer cell targeting
How TAT-GNS are released and target cancer cells are key issues that can determine the delivery efficiency and therapeutic effect of the TAT-GNS loaded MSC. The supernatant of TAT-MSC was collected and characterized before and after laser irradiation. The TAT-GNS loaded MSCs could excrete GNS clusters via exocytosis as shown by TEM (Fig. 4A). Furthermore, a thin layer of membrane was found on the surface of the excreted GNS clusters (Fig. 4B). The size of the excreted GNS was determined by DLS and the average size was around 653 nm (Fig. 4C), which was consistent in four independent samples (Fig. S7) and released in a time dependent manner (Fig. S8). A red-shift of the plasmon resonance was also observed in the UV-Vis-NIR spectrum of the GNS-loaded MSCs supernatant with a maximum absorbance at 810 nm (Fig. 4D). The membrane content on the excreted GNS clusters was determined by SDS-PAGE gel electrophoresis (Fig. 4E). And the protein of the MVs@GNS were almost the same as the control group. The excreted GNS clusters collected from GNS-loaded MSCs displayed a same protein profile as the MSC extracellular microvesicles. It demonstrated that the proteins containing in the microvesicles almost the same as the cell membrane, but some proteins such as the stripe of 20 kDa had different expression levels (Fig. S9). It indicates that the excreted GNS clusters could be encapsulated in the microvesicles, which could selectively deliver the nanoparticles into the cancer cells. More importantly, the release of GNS clusters could be further facilitated via laser exposure at 808 nm, as indicated by the increased amount of black dots in the extracellular supernatant after irradiation (Fig. S10). The protein quantification further confirmed that the laser treatment could enhance 35% protein release in the supernatant (Fig. 4F). It suggests that the light could be the external trigger to control the GNS release from the carrier cells. And the protein release in high power NIR (2.5 W/cm2) was much lower, which was due to the cell death under the NIR exposure to inhibit the microvesicles' release (Fig. S11 and Fig. S12). The activated caspase 3 staining result showed that the death was not though the apoptotic pathway (Fig. S12C).
Microvesicles containing GNS clusters released from GNS-loaded MSCs and their transportation to cancer cells. A. Representative images showing an intracellular GNS cluster (blue arrow) and GNS clusters exocytosis (red arrow) by GNS-loaded MSCs. B. GNS clusters produced by exocytosis from GNS-loaded MSCs in the extracellular medium. C. Size distribution of the extracellular microvesicles containing GNS clusters by DLS. D. UV-Vis-NIR spectrum of microvesicles containing GNS clusters in PBS. E. SDS-PAGE protein analysis of microvesicles containing GNS clusters. Samples were stained with Coomassie brilliant blue. I, markers. II, MSCs extracellular vesicles as control. III, GNS-loaded MSCs extracellular vesicles. F. Protein quantification of supernatant in GNS loaded MSCs by BCA assay 4 h after NIR exposure (1.5 W/cm2, 3 min). The concentration of TAT-GNS was 160 pM. G. Confocal laser scanning microscopy of FITC-labeled GNS clusters released from MSCs to PC-3 (labeled by RFP). The red arrow shows GNS clusters labeled by FITC and taken up by a PC-3 cell (labeled by RFP).
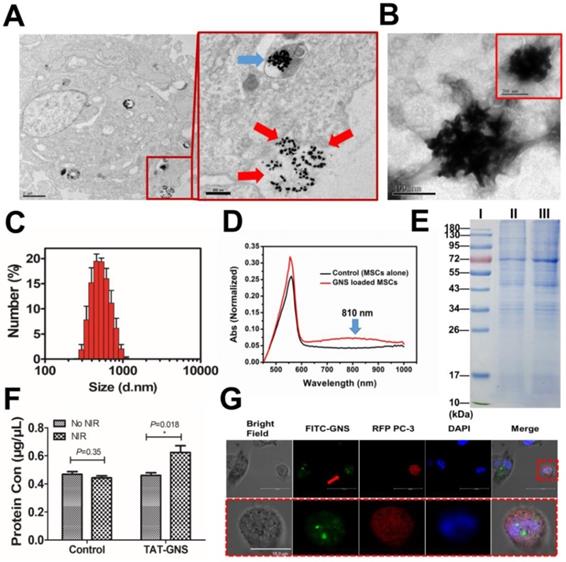
The microvesicles released from cells are natural carriers that can transport compounds to the targeted cells.39-44 To illustrate the delivery mechanism of the excreted GNS loaded microvesicles, the GNS and PC-3 cells were labeled with FITC and red fluorescence protein (RFP), respectively. The MSCs loaded with FITC-TAT-GNS were co-cultured with RFP labeled PC-3 cells at 1:1 ratio (Fig. 4G). After 24h, the FITC-TAT-GNS signal could be observed in the PC-3 cells, indicating the transportation of GNS loaded microvesicles into PC-3 cells (Fig. 4G and Fig. S13). The isolated microvesicles containing FITC-TAT-GNS (MVs@FITC-GNS) could also be uptake by the PC-3 cells (Fig. S14). To determining the tumor targeting ability, the mouse embryonic fibroblast 3T3 cells was used as the control cell line. The supernatant of MSCs@FITC-TAT-GNS was collected after 24 h and added in the 3T3 cells and PC-3 cells by a 1:1 ratio with normal medium. The fluorescence in the supernatant of MSCs@FITC-TAT-GNS indicated that the FITC-TAT-GNS could be released into the culture medium (Fig. S15). The confocal imaging indicated that MVs@FITC-GNS could be uptake by PC-3 cells but the majority of the MVs@FITC-GNS were localized outside of the 3T3 cells (Fig. S16 & Fig. S17). The results indicate that the GNS cluster loaded microvesicles may retain the tumor targeting ability, which is similar as the MSC extracellular vesicles.38, 60
In vitro PTT effect
The PTT efficacy of the TAT-GNS loaded MSCs was evaluated to release the nanoparticles and prevent the risk of tumorigenesis by stem cells (Fig. 5). The MSCs were incubated with 0, 20, 40, 80 or 160 pM TAT-GNS for 24 h. The live/dead cell staining was performed in MSCs 4 h after exposing to an 808 nm laser (optical density 2.5 W/cm2, 3 min). It was found that TAT-GNS started to show good cytotoxicity effect to MSCs at 40 pM TAT-GNS incubation condition, indicating by the red fluorescence of cells from PI staining (Fig. 5A). Complementarily, trypan blue staining assay showed similar destruction and further confirmed the PTT effect (Fig. S18). Up to 55.6 % MSCs were dead after irradiation quantified by the CCK8 assay (Fig. 5C). In addition, the PTT effect could be further enhanced via increasing the TAT-GNS concentration. Notably, majority of the MSCs could be damaged with the incubation of 80 and 160 pM TAT-GNS after laser exposure (Fig. 5A and Fig. 5C). It indicates that the MSCs could perform a “suicide bomber”-like function and reduce the risk of tumorigenesis.
In vitro PTT effect of GNS-loaded MSCs. A. PTT effects on GNS-loaded MSCs. B. Photothermal therapy effects on co-cultured GNS-loaded MSCs and PC-3 with different ratios (ranging from 1:4 to 4:1). Representative 10× images obtained 4 hours after laser exposure (Live-dead staining with PI and calcein-AM); C. Cell viability of GNS-loaded MSCs post light irradiation; D. Cell viability of co-cultured GNS-loaded MSCs and PC-3 post PTT. Error bars indicate s.d. (n=4). P < 0.05(*), P < 0.01(**), P < 0.001 (***) compared with the control group.
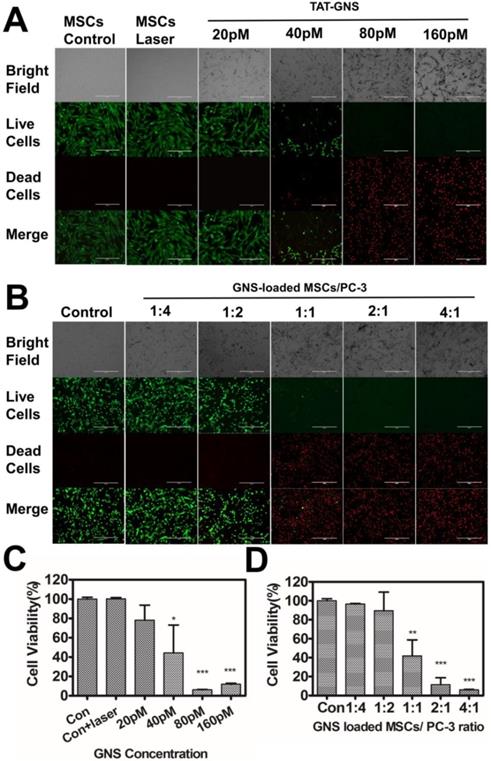
Subsequently, the PTT effect on prostate cancer cells were determined by co-cultured with TAT-GNS loaded MSCs with a series of ratios. The MSCs were pre-incubated with 160 pM TAT-GNS for 24 h. The co-culture ratio was ranged from 1:4 to 4:1 (MSCs/PC-3 cells) and the cell viability was determined by CCK-8 assay. It was found that all cells were alive indicated by the green color of Calcein after co-culturing at low ratios of MSCs/PC-3 cells (1:4 and 1:2) after laser irradiation. In contrast, when the co-cultured ratio of MSCs/PC-3 cells increased to 1:1, 2:1 and 4:1, the amounts of dead cells (in red color) were significantly increased after light exposure (Fig. 5B). The dead cells increased up to 58.1 % at the co-cultured ratio of 1:1 (Fig. 5D). And at 2:1 and 4:1 ratio, over 90 % of the cancer cells could be eradicated upon PTT. It indicates that the GNS-loaded MSCs could effectively damage cancer cells via photothermal treatment (Fig. 5D).
MSCs improved the intratumoral GNS distribution and PTT efficacy in vivo via intratumoral injection
The excellent in vitro results promote us to investigate the intratumoral distribution and PTT effect on the animal model. PC-3 prostate cancer cells were implanted in the flank of mice. When the volumes of the tumor increased upon 62.5 mm3, the mice were randomized into three treatment groups. Each group (n = 5) received intratumoral injections of phosphate buffered saline (PBS), free TAT-GNS, or GNS-loaded MSCs. To test whether MSCs-mediated delivery of GNS could improve the distribution in tumors, photoacoustic imaging was utilized to trace the GNS in vivo post 3 days of injection (Fig. 6A). The GNS signals were observed in both of the GNS and GNS-loaded MSCs treated groups (Fig. 6A). The tumor injected with TAT-GNS alone showed the localized signal spot with the area of 0.022 cm2. In contrast, GNS-loaded MSCs showed a relative even distribution of the nanoparticles in the entire tumor with the area of 0.073 cm2. The histology analysis was carried out to further investigate the GNS-loaded MSC delivery. Post 3 days of injection, the tumors were collected for H&E and silver co-staining. The GNS (as shown in black and brownish color) in the GNS treated group were mainly localized at the injection site (Fig. 6B). No GNS could not be found at the tumor border. By contrast, the GNS-loaded MSCs showed broad distribution of the GNS in either tumor border or interior (noted by yellow arrows), which was consistent with the photoacoustic imaging results. It confirms that MSCs could have the migration ability to target the cancer cells in vivo.
Photothermal ablation of prostate tumors via GNS-loaded MSCs. A. PAI of GNS distribution after intratumoral injection. Fusion of ultrasound and PAI images of the tumors after the injection of TAT-GNS and GNS-loaded MSCs. Scale bar is 0.5 cm. B. Representative H&E and silver staining sections of the tumor after PA imaging. The yellow circles and yellow arrows noted the GNS signals (brown spots) in the sections. All scale bars are 200 μm. C. Infrared microscopic imaging: NIR laser irradiation of the tumor bearing mice after injections of PBS, TAT-GNS and GNS-loaded MSCs after 3 days. D. Temperature rise profiles at the tumor site under the 808 nm NIR laser irradiation with an intensity of 1 W/cm2 for 10 min. E. Relative tumor volume from mice intratumorally injected with PBS, GNS and GNS-loaded MSCs (n = 5) post PTT; F. Representative images of PC-3 tumors harvested from the three different groups post 16 days of laser irradiation. G. Representative H&E staining section of the mice organs after treatment for 16 days. All scale bars are 200 μm.
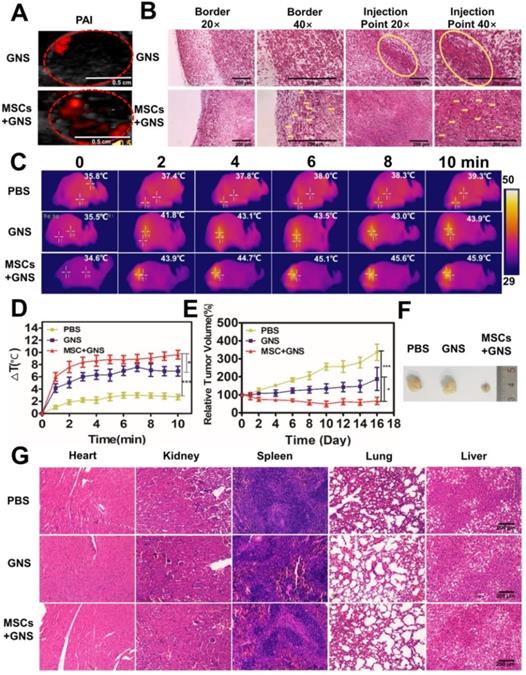
After 3 days of injection, three treatment groups (each group n = 5) were anesthetized and all tumors were exposed to an 808 nm NIR light (1 W/cm2, 10 min). The GNS-loaded MSCs-treated group showed good photothermal response and the temperature at the tumor area could rapidly increase upon irradiation (Fig. 6C and Fig. 6D). After 2 min exposure, 43.9 ºC was reached at the tumor area in the GNS-loaded MSCs-treated group, which was higher than the PBS (37.4 ºC) and GNS (41.8 ºC) treated groups (Fig. 6C and Fig. 6D). The temperature could further increase with prolonged irradiation time (Fig. 6C and Fig. 6D). With the good photothermal effect and broad intratumoral GNS distribution, GNS-loaded MSC treated group had an effective tumor inhibition effect after PTT (Fig. 6D and 6E). The PBS treated group showed a continuous tumor growth after irradiation, indicating the light had little inhibition effect to tumors (Fig. 6D and 6E). The GNS treated group had a moderate PTT effect as shown by the slowly increased tumor volume. Compared with the PBS and GNS treated groups, the GNS-loaded MSC group showed the best tumor inhibition effect. The tumor growth was completely inhibited and the tumor volume decreased post 16 days of PTT (Fig. 6D and 6E). In addition, the body weights of mice were normal during the experimental period (Fig. S19). No severe side effects were observed during and post treatment. Compared to the gold nanoparticle alone, the GNS loaded MSCs obtained a superior tumor suppressive effect with a wider GNS intratumor distribution via the single injection. It should be noted that the ratio of TAT-GNS loaded MSCs to prostate cancer cells appears to be much lower in the tumor tissue in vivo (Fig. 6B) compared with that in vitro. As shown in Fig. 5B and 5D, at least 1:1 MSC/cancer cell ratio should be utilized for TAT-GNS loaded MSCs mediated cancer cell killing. Significant anti-tumor growth was observed in vivo at only 105 cells TAT-GNS loaded MSCs per mouse, suggesting better heat preservation effect in the animal body compared with the two-dimensional cell culture environment.
The histology analysis was carried out to further investigate the GNS-loaded MSC delivery and PTT effect. Post 3 days of injection, the tumors were collected for H&E and silver co-staining. The GNS (as shown in black and brownish color) in the GNS treated group were mainly localized at the injection site (Fig. 6F). No GNS could not be found at the tumor border. By contrast, the GNS-loaded MSCs showed broad distribution of the GNS in either tumor border or interior (noted by yellow arrows), which was consistent with the photoacoustic imaging results. It confirms that MSCs could have the migration ability to target the cancer cells in vivo. In addition, tumors and organs were collected after 16 days of therapy (Fig. 6G and Fig. S20). In the GNS treated group, the GNS were located near the needle path and induced the necrosis in the injection area (as shown by red arrows in Fig. S20). In the GNS-loaded MSC group, majority of GNS were found in the scar tissues (Fig. S20), which indicated that the wider distribution of GNS could induce necrosis under the NIR light exposure. No significant tissue damage was observed in the organ tissues post PTT (Fig. 6G), indicating the local PTT therapy had the great selectivity and slight side effects.
It should be noted that effective accumulation and distribution of gold nanoparticles in tumors are crucial for gold nanoparticles-mediated PTT. El-Sayed et al. found that the intratumoral injection of gold nanoparticles was more effective than intravenous injection.61 Although gold nanoparticles showed a better accumulation in tumors by intratumoral injection, they lacked a good distribution in the tumor area. The insufficient diffusion ability of nanoparticles limits the homogenous temperature rise in the entire tumor which in turn could hamper the treatment efficiency.31, 62 In our platform, using the MSCs as vehicles, the targeting ability of MSCs mediated by SDF-1/CXCR4 can improve the tumor selectivity through the concentration gradient of SDF-1 secreted by tumor cells.63-65 The MSCs act as a “smart vehicle” to assemble and delivery nanoparticles with high GNS loading capacity and retain “homing” effect to cancer cells. Both of the MSCs and their secreted microvesicles could facility the tumor targeting ability for the GNS. Our results indicate that the intratumoral injection of the MSCs could facilitate the GNS distribution in the tumor area which leads to the excellent PTT effect in vivo. Since the targeting approach by tumor-tropic cells is based on multiple targeting mechanisms, it may be more effective than a single targeting molecule or antibody modification. Furthermore, the combination of GNS with MSCs could prevent the teratoma formation, which is a major concern of stem cell transplantation.63 The GNS-loaded MSCs could act as a “Trojan Horse” controlled by NIR light and minimize the risk of tumorigenesis post intratumoral injection. After laser exposure, the stem cells can be damaged by the hyperthermia and release the microvesicles containing GNS clusters for cancer cell targeting and uptake.
MSCs improved the anti-tumor PTT efficacy in vivo via intravenous injection
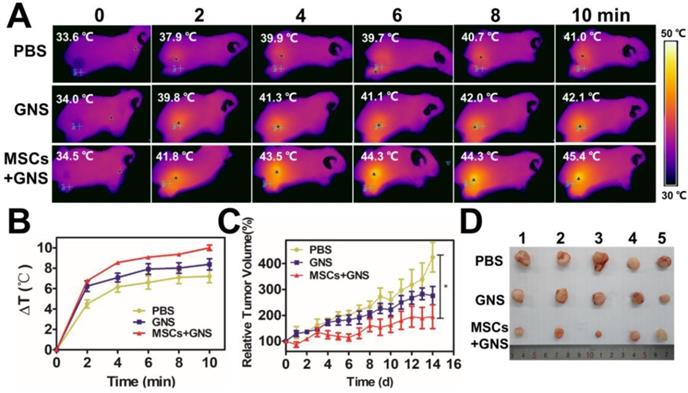
As the conspicuous effect in the local administration, the anti-tumor effect of GNS-loaded MSCs in vivo was further determined by intravenous injection. Each group (n = 5) received a NIR light exposure (1.5 W/cm2 for 10 min) 3 days after intravenous injection with PBS, TAT-GNS, GNS-loaded MSCs. The GNS-loaded MSCs group showed higher temperature increasing at the tumor area that could reach to 43.9 ± 1.0 °C (Fig. 7A and Fig. 7B). It indicated that the MSCs could improve the GNS accumulation in the tumor area by systemic delivery. After 14 days after NIR treatment, the body weights of mice were all normal (Fig. S21). The GNS-loaded MSC treated group showed an effective tumor inhibition effect after PTT compared with PBS group (P < 0.05) in 14 days (Fig. 7C and Fig. 7D). The “homing effect” of MSCs intravenous injection were determined by ex vivo fluorescence image. The fluorescence signals were observed most in the mouse lungs and tumors 3 days after injection (Fig. S22). It confirmed that the GNS-loaded MSCs could maintain the tumor tropic migration ability after loading GNS in vivo with the migration results in vitro (Fig. 2E). Although it showed good anti-tumor effect in intravenous administration, the MSCs would be trapped mostly in the pulmonary capillary by pulmonary circulation. The administration approaches should be considered according to the disease specificity in the further applications.
Photothermal ablation of prostate tumors via intravenous injection. A. Infrared microscopic imaging, NIR laser irradiation (808 nm, 1.5 W/cm2, 10 min) of the tumor bearing mice after intravenous injections of PBS, TAT-GNS and GNS-loaded MSCs (5 × 105 cells) after 3 days. B. Temperature rise profiles at the tumor site under the 808 nm NIR laser irradiation with an intensity of 1.5 W/cm2 for 10 min. C. Relative tumor volume from mice intravenously injected with PBS, TAT-GNS and GNS-loaded MSCs (n = 5) post PTT in 14 days; D. Representative images of PC-3 tumors harvested from the three different groups post 14 days of laser irradiation.
Conclusion
In summary, our work shows an effective delivery platform utilizing MSCs and the extracellular microvesicles containing assembled GNS for targeted PTT of prostate cancer with tumorigenesis exemption in vitro and in vivo. The conjugated TAT peptide on the surface of GNS could enhance cellular uptake and improve loading efficiency. Importantly, the stem cells act not only as the cellular carrier to load GNS into the lysosomes but also the “cellular engineering factory” to assemble the GNS and encapsulate them into microvesicles under the light control. The MSCs loaded with TAT-GNS maintained “homing” effect and tropic migration ability to improve the GNS intratumoral distribution and PTT effect in vivo both local injection and systemic delivery. To the best of our knowledge, it is the first example of utilizing both of MSCs and their excreted microvesicles as an intelligent “Trojan Horse” to assemble and transport the nanoparticles for targeted cancer treatment. We believe that our work presents a comprehensive understanding of the MSCs-based nanoparticle transportation and hold the promise as an effective strategy for cancer therapy including prostate cancer. Further studies on active self-assembly of nanoparticles inside the cells will be carried out in the future.
Experimental Section
Materials
Chloroauric acid (HAuCl4) was purchased from Civi-Chem (Suzhou, China). HS-PEG-COOH (MW = 5,000) were purchased from Pengshuo Biotech Co., Ltd. (Shanghai, China). Silver nitrate (AgNO3) was purchased from Alfa (America). Ascorbic acid (AA) was purchased from Macklin (Shanghai, China). Hydrochloric acid (HCl) was purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China).1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-Hydroxysuccinimide (NHS) were purchased from Aladdin (Shanghai, China). Phosphate buffer saline (PBS), high glucose Dulbecco's Modified Eagle's Medium (DMEM) were purchased from HyClone (America). Penicillin/ streptomycin and fetal bovine serum (FBS) were purchased from Gibco (America). Basic fibroblast growth factor (bFGF) was purchased from Invitrogen (America). TAT-peptide (residues 47-57 sequence YGRKKRRQRRR-NH2) and FITC labeled TAT were purchased from GL Biochem Ltd (Shanghai, China). Cell counting kit-8 (CCK-8) was purchased from Beijing Zoman Biotechnology Co., Ltd. Propidium Iodide (PI) stain, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), Western and IP kit was purchased from Beyotime Biotechnology (Shanghai, China). Culture-Inserts were purchased from Ibidi (Germany).
Cell lines and culture
The PC-3, DU145, LNCaP, 3T3 cells were cultured in high glucose Dulbecco's Modified Eagle Medium (DMEM) containing 10 % fetal bovine serum (FBS) and 1 % penicillin-streptomycin (Gibco). The MSCs were collected from the human umbilical cord and cultured in low glucose DMEM with 10 % FBS and 10 ng/mL basic Fibroblast Growth Factor (bFGF, Invitrogen). All cells lines were maintained in a humidified atmosphere at 37°C with 5 % CO2/95 % air. All of the cells were passaged using 0.25 % trypsin when the cells reached 70 %-90 % confluence.
Synthesis and characterization of TAT-GNS
The GNS were synthesized through a seed-mediated growth approach46. The seed was obtained by vigorous mixing sodium citrate (44.6 mg) in HAuCl4 solution (1 mM) under boiling temperature. After 15 min reaction, the solution was wine red and cooled to room temperature. The seed solution was purified by filtration through a 0.22 μm nitrocellulose membrane and kept at 4 °C for long-term storage. For GNS synthesis, the seed solution (600 μL) was added to of HAuCl4 solution (0.25 mM, 60 mL) with HCl (0.01 M) in a 100 mL glass vial at room temperature under moderate stirring (700 rpm). AgNO3 (3 mM, 600 μL) and AA (100 mM, 300 μL) were mixed quickly in the above solution. After stirring for 30 s, the solution color rapidly turned from light red to greenish-black indicating the formation of the GNS. Then, HS-PEG-COOH (MW 5000, 5 mg/mL, 2 mL) was immediately added into the solution under gentle stirring overnight. The PEGylated GNS was purified by centrifugation at 9,500g for 15min and then resuspended in double-distilled water. TAT-GNS was prepared by mixing TAT-peptide (30 µM) in PEGylated nanostars (0.3 nM) for 24 hours under EDC/NHS (25 μmol EDC and 30 μmol NHS) coupling reaction condition in a 10 mL system. Then the TAT-GNS was purified by centrifugation at 9,500g for 15min three times. The DLS and zeta potential of bare GNS, PEG-GNS, TAT-GNS were determined by Malvern Zetasizer Nano ZS90. The near infrared absorption was determined by UV-Vis spectroscopy (Cary 60 UV-Vis, Agilent Technologies). The infrared spectrum was determined by FTIR spectroscopy (Cary 630 FTIR, Agilent Technologies).
Cell viability of MSCs and cellular uptake of TAT-GNS
Cell viability was determined by CCK8 assay. The huMSCs cells were seeded in 96-well plates with 5,000 cells per well in culture medium. After overnight incubation, the cells were treated with TAT-GNS of different concentrations (ranging from 20 pM to 160 pM) when the confluency of the cells came up to 70 %. After 24 h incubation at 37 °C, 10 μL of CCK-8 solution was added to each well. After 2 h incubation at 37 °C, absorbance at 450 nm was read on a microplate reader (ELx808, BioTek). 4 replicate wells were used for each condition. Cellular uptake was determined by microscopic observation, the huMSCs cells and PC-3 cells were seeded in 96-well plates at 5000 cells/well in culture medium. After overnight incubation, the cells were treated with TAT-GNS of different concentration (range from 20 pM to 160 pM). After 24 h incubation at 37 °C, the cellular uptake of GNS was observed using the light microscope.
In vitro migration assay
The migration capability of GNS-loaded MSCs was determined by the wound healing assay. The culture-inserts were used for this study. The MSCs were seeded in the left well 70 μL per well with the concentration of 5×105 cells/mL, PC-3 cells were planted in the right well with the same concentration. And the MSCs were incubated with 0, 80 pM, 160 pM TAT-GNS for 24 h. Then the culture-inserts were removed and the gaps between MSCs and PC-3 cells were maintained. The same procedure was used to determine the migration capability of GNS-loaded MSCs for DU145 and LNCaP prostate cancer cell lines.
Investigation of in vitro photothermal effect of TAT-GNS
The MSCs were incubated with TAT-GNS by different concentration (ranging from 0-160 pM) for 24 h in 96-well plates. The plates were carefully washed three times with PBS to remove the unloaded TAT-GNS. The GNS-loaded MSCs cells were irradiated by continuous-wave 808 nm laser (ANJ, Beijing, China) with an optical density of 2.5 W/cm2 for 3 min. The co-staining of Calcein AM (green, staining 30 min) and PI (red, staining 10 min) was used to differentiate live and dead cells at 4 hours after in vitro PTT. The results were observed using the fluorescence microscope (EVOS FL Auto, life technologies). Subsequently, the cell viability was determined by CCK-8 assay. The localized photothermal effect in vitro of GNS-loaded MSCs with co-cultured PC-3 cells was further investigated with different ratios (ranging from 1:4 to 4:1). The plate was irradiated by 808 nm laser with an optical density of 2.5 W/cm2 for 3 min. The cell death was observed utilizing co-staining of Calcein AM and PI. And the cell viability was quantified by CCK-8 assay.
Flow cytometry assay of the surface markers of GNS-loaded MSCs
The MSCs were incubated with TAT-GNS (160 pM) for 24 h in the 6-well plates. The both GNS-loaded and unloaded MSCs were tested the expression of positive surface markers for CD29, CD44, CD90, CD105 and negative surface markers for CD45 and CD34 by BD FACSCalibur. Briefly, 1×105 cells from each sample were washed two times with PBS containing 1 % BSA and then were incubated with the following fluorophore-conjugated monoclonal antibodies: CD44-V450, CD105-PerCP-Cy5.5, CD45-PE, CD34-PE, CD90-FITC and CD29-APC for 30 min in a dark room. Then the cells were washed with PBS containing 1 % BSA and centrifuged and were evaluated using BD FACSCalibur.
CXCR4 expression of GNS-loaded MSCs
The MSCs were seeded in 6 well plates (1×105 per well), and treated with 0, 40, 80, 160 pM TAT-GNS for 24 h and washed twice by PBS. After collection of cells, the total proteins expressed in cells were separated by a Cell lysis buffer for Western and IP kit (Beyotime). And the CXCR4 expression of MSCs was performed by Western blot.57 And 30 μg protein was loaded per lane for the Western blot.
TEM observation for cellular assemble for GNS in MSCs
The MSCs were seeded in the 6-well plates (1×105 per well), and treated with TAT-GNS (160 pM) for 4 h and 24 h, respectively. The cells were washed twice then collected through centrifugation at 1000 rpm for 3min. Then the samples were fixed with 2.5 % glutaraldehyde at 4 °C overnight and followed by 1 % aqueous osmium tetroxide staining. TEM images were performed under 300 kV in a FEI Tecnai F30 microscope equipped with a Gatan CCD digital camera.
Microvesicles separation and collection
The MSCs were treated with 160 pM TAT-GNS for 24 h in the 6-well plates (1×105 per well). And the cells were washed twice before incubated with serum-free medium. The supernatant was collected after 24 h and replaced by fresh serum-free medium in 2 days. The collected medium was centrifuged (700 g for 10 min) to eliminate suspending cells and large apoptotic bodies. Then the medium was centrifuged (150,000 g for 1.0 h) and the GNS loaded microvesicles were collected for further detection.
The establishment of animal models
All animals were treated in accordance with the guidelines of the animal care and use committee at Tongji University. Male nude mice were purchased and bred at the Center of Experimental Animals at Tongji University. To prepare the prostate subcutaneous tumor model, male nude mice were anesthetized by intraperitoneal injection with 10 % chloral hydrate. Subsequently, PC-3 tumor tissue block (1 mm3) were implanted into the right groin of each mouse.
In vivo photoacoustic imaging
When the subcutaneous tumor volume increased upon 62.5 mm3, six nude mice were received intratumoral injection of free TAT-GNS (43.73 μg, 10 μL), or GNS-loaded MSCs (43.73 μg of GNS in 1×105 MSCs, 10 μL), three mice in each group. The number of MSCs was determined by cell counting chamber. After 3 days, the GNS distribution in tumor was determined via the photoacoustic imaging (780 nm, 40 mJ/cm2).
In vivo PTT by intratumoral injections
When the volumes of the tumors increased upon 62.5 mm3 (the diameters of the tumors increased upon 5 mm), the mice were randomized into three treatment groups. Each group (n = 5) received intratumoral injections of either phosphate buffered saline (PBS, 10 μL), free TAT-GNS (43.73 μg, 10 μL), or GNS-loaded MSCs (43.73 μg of GNS in 1×105 MSCs, 10 μL). After 3 days, mice were anesthetized and all tumors were exposed to an 808 nm NIR laser (continuous-watt laser) with a power of 1 W/cm2 for 10 min. The body weight and tumor volume were monitored every other day. Tumor volumes were measured using a caliper every other day, and tumor volume was calculated by Volume (mm3) = L*W2/2, where L is the longest dimension and W is measured perpendicular to L.
In vivo PTT and fluorescence imagings of MSCs by intravenous injection
Each group (n = 5) received intravenous injections of either phosphate buffered saline (PBS, 200 μL), free TAT-GNS (218.65 μg, 200 μL), or GNS-loaded MSCs (218.65 μg of GNS in 5×105 MSCs, 200 μL). After 3 days, all the mice were received a NIR light exposure (1.5 W/cm2 for 10 min). The body weight and tumor volume were monitored every day. The MSCs and GNS-loaded MSCs were labeled with DiI (10 mM) 20 min and washed with PBS three times. The PC-3 bearing nude mice were intravenously injected with 5×105 DiI labeled MSCs and GNS-loaded MSCs each mouse. The ex vivo fluorescence images were determined 3 days after intravenous injection using a Berthold NightOWL LB 983 In Vivo Imaging System (Bad Wildbad, Germany).
Statistical analysis
Data were shown as means ± SD. Statistical analyses were carried out by Student's t test with SPSS software for windows version 19.0 (SPSS Inc, Chicago, IL, USA). P Values at P < 0.05(*), P < 0.01(**), P < 0.001 (***) were considered as statistically significant.
Acknowledgements
This work was financed by National Science Foundation of China (No.81571803). Y.C. thanks the Thousand Talents Plan, Shanghai Municipal Education Commission Innovative Program (No.2017-01-07-00-07-E00038), and Shanghai Science and International Cooperation Program (No.16410724300). X.F.W. thanks the Special Diseases Program of Pudong New Area Health System (No. PWZzb2017-06). L.Q.H thanks the Fundamental Research Funds for the Central Universities (No. 22120180030). We thank Yimei Chen at the Electron Microscopy Core Facility of the University of Chicago and Yajing Shen at the Institute for Translational Nanomedicine of Shanghai East Hospital for the assistance in TEM image analysis. We thank Lei Gao at the School of Medicine Tongji University for the assistance in SDS-PAGE protein analysis, Chuyi Zhang at the School of Medicine Tongji University for the assistance in Western-blot protein analysis, Prof. Jian Zhao and Dr. Guangxue Wang at the Shanghai East Hospital for the cell studies.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Heidenreich A, Bastian PJ, Bellmunt J. et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014;65:467-79
3. Penson DF, McLerran D, Feng Z. et al. 5-year urinary and sexual outcomes after radical prostatectomy: results from the Prostate Cancer Outcomes Study. J Urol. 2008;179(5 Suppl):S40-4
4. Valerio M, Ahmed HU, Emberton M. et al. The role of focal therapy in the management of localised prostate cancer: a systematic review. Eur Urol. 2014;66:732-51
5. Han J, Li J, Jia W. et al. Photothermal therapy of cancer cells using novel hollow gold nanoflowers. Int. J. Nanomed. 2014;9:517-26
6. Ma Y, Tong S, Bao G. et al. Indocyanine green loaded SPIO nanoparticles with phospholipid-PEG coating for dual-modal imaging and photothermal therapy. Biomaterials. 2013;34:7706-14
7. Robinson JT, Tabakman SM, Liang Y. et al. Ultrasmall reduced graphene oxide with high near-infrared absorbance for photothermal therapy. J. Am. Chem. Soc. 2011;133:6825-31
8. Yang K, Zhang S, Zhang G. et al. Graphene in mice: ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano lett. 2010;10:3318-23
9. Yang W, Guo W, Le W. et al. Albumin-Bioinspired Gd:CuS Nanotheranostic Agent for In Vivo Photoacoustic/Magnetic Resonance Imaging-Guided Tumor-Targeted Photothermal Therapy. ACS Nano. 2016;10:10245-57
10. Hwang S, Nam J, Jung S. et al. Gold nanoparticle-mediated photothermal therapy: current status and future perspective. Nanomedicine. 2014;9:2003-22
11. Robinson R, Gerlach W, Ghandehari H. Comparative effect of gold nanorods and nanocages for prostate tumor hyperthermia. J. Controlled Release. 2015;220:245-252
12. Stern JM, Kibanov Solomonov VV, Sazykina E. et al. Initial Evaluation of the Safety of Nanoshell-Directed Photothermal Therapy in the Treatment of Prostate Disease. Int. J. Toxicol. 2016;35:38-46
13. Gad SC, Sharp KL, Montgomery C. et al. Evaluation of the toxicity of intravenous delivery of auroshell particles (gold-silica nanoshells). Int. J. Toxicol. 2012;31:584-94
14. O'Neal DP, Hirsch LR, Halas NJ. et al. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209:171-6
15. Fay BL, Melamed JR, Day ES. Nanoshell-mediated photothermal therapy can enhance chemotherapy in inflammatory breast cancer cells. Int. J. Nanomed. 2015;10:6931-41
16. Day ES, Thompson PA, Zhang L. et al. Nanoshell-mediated photothermal therapy improves survival in a murine glioma model. J Neuro-Oncol. 2011;104:55-63
17. Comenge J, Fragueiro O, Sharkey J. et al. Preventing Plasmon Coupling between Gold Nanorods Improves the Sensitivity of Photoacoustic Detection of Labeled Stem Cells in Vivo. ACS Nano. 2016;10:7106-16
18. Wang D, Wang T, Xu Z. et al. Cooperative Treatment of Metastatic Breast Cancer Using Host-Guest Nanoplatform Coloaded with Docetaxel and siRNA. Small. 2016;12:488-98
19. Camposeo A, Persano L, Manco R. et al. Metal-Enhanced Near-Infrared Fluorescence by Micropatterned Gold Nanocages. ACS Nano. 2015;9:10047-54
20. Wang S, Tian Y, Tian W. et al. Selectively Sensitizing Malignant Cells to Photothermal Therapy Using a CD44-Targeting Heat Shock Protein 72 Depletion Nanosystem. ACS Nano. 2016;10:8578-90
21. Wei P, Chen J, Hu Y. et al. Dendrimer-Stabilized Gold Nanostars as a Multifunctional Theranostic Nanoplatform for CT Imaging, Photothermal Therapy, and Gene Silencing of Tumors. Adv Healthc Mater. 2016;5:3203-3213
22. Li N, Zhao P, Astruc D. Anisotropic gold nanoparticles: synthesis, properties, applications, and toxicity. Angew. Chem, Int. Ed. Engl. 2014;53:1756-89
23. Nehl CL, Liao H, Hafner JH. Optical properties of star-shaped gold nanoparticles. Nano lett. 2006;6:683-8
24. Hrelescu C, Sau TK, Rogach AL. et al. Single gold nanostars enhance Raman scattering. Appl. Phys. Lett. 2009;94:153113
25. Liu Y, Ashton JR, Moding EJ. et al. A Plasmonic Gold Nanostar Theranostic Probe for In Vivo Tumor Imaging and Photothermal Therapy. Theranostics. 2015;5:946-60
26. Mortimer GM, Butcher NJ, Musumeci AW. et al. Cryptic epitopes of albumin determine mononuclear phagocyte system clearance of nanomaterials. ACS Nano. 2014;8:3357-66
27. Yang B, Han X, Ji B. et al. Competition Between Tumor and Mononuclear Phagocyte System Causing the Low Tumor Distribution of Nanoparticles and Strategies to Improve Tumor Accumulation. Curr. Drug Delivery. 2016;13:1261-1274
28. Madsen SJ, Baek SK, Makkouk AR. et al. Macrophages as cell-based delivery systems for nanoshells in photothermal therapy. Ann Biomed Eng. 2012;40:507-15
29. Madsen SJ, Christie C, Hong SJ. et al. Nanoparticle-loaded macrophage-mediated photothermal therapy: potential for glioma treatment. Lasers Med Sci. 2015;30:1357-65
30. Rao L, Bu LL, Ma L. et al. Platelet-Facilitated Photothermal Therapy of Head and Neck Squamous Cell Carcinoma. Angew. Chem, Int. Ed. Engl. 2018;57:986-991
31. Mooney R, Roma L, Zhao D. et al. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS Nano. 2014;8:12450-60
32. Cheng Y, Morshed R, Cheng SH. et al. Nanoparticle-programmed self-destructive neural stem cells for glioblastoma targeting and therapy. Small. 2013;9:4123-9
33. Liu Y, Yang M, Zhang J. et al. Human Induced Pluripotent Stem Cells for Tumor Targeted Delivery of Gold Nanorods and Enhanced Photothermal Therapy. ACS Nano. 2016;10:2375-85
34. Yang M, Liu Y, Hou W. et al. Mitomycin C-treated human-induced pluripotent stem cells as a safe delivery system of gold nanorods for targeted photothermal therapy of gastric cancer. Nanoscale. 2017;9:334-340
35. Kang S, Bhang SH, Hwang S. et al. Mesenchymal Stem Cells Aggregate and Deliver Gold Nanoparticles to Tumors for Photothermal Therapy. ACS Nano. 2015;9:9678-90
36. Wu J, Liu Y, Tang Y. et al. Synergistic Chemo-Photothermal Therapy of Breast Cancer by Mesenchymal Stem Cell-Encapsulated Yolk-Shell GNR@HPMO-PTX Nanospheres. ACS Appl Mater Interfaces. 2016;8:17927-35
37. Nakki S, Martinez JO, Evangelopoulos M. et al. Chlorin e6 Functionalized Theranostic Multistage Nanovectors Transported by Stem Cells for Effective Photodynamic Therapy. ACS Appl Mater Interfaces. 2017;9:23441-9
38. Yeo RW, Lai RC, Zhang B. et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv. Drug Delivery Rev. 2013;65:336-41
39. Wilhelm C, Lavialle F, Pechoux C. et al. Intracellular trafficking of magnetic nanoparticles to design multifunctional biovesicles. Small. 2008;4:577-82
40. Silva AK, Wilhelm C, Kolosnjaj-Tabi J. et al. Cellular transfer of magnetic nanoparticles via cell microvesicles: impact on cell tracking by magnetic resonance imaging. Pharm. Res. 2012;29:1392-403
41. Al Faraj A, Gazeau F, Wilhelm C. et al. Endothelial cell-derived microparticles loaded with iron oxide nanoparticles: feasibility of MR imaging monitoring in mice. Radiology. 2012;263:169-78
42. Silva AK, Di Corato R, Pellegrino T. et al. Cell-derived vesicles as a bioplatform for the encapsulation of theranostic nanomaterials. Nanoscale. 2013;5:11374-84
43. Silva AK, Kolosnjaj-Tabi J Bonneau S. et al. Magnetic and photoresponsive theranosomes: translating cell-released vesicles into smart nanovectors for cancer therapy. ACS Nano. 2013;7:4954-66
44. Armstrong JP, Holme MN, Stevens MM. Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano. 2017;11:69-83
45. Betzer O, Perets N, Angel A. et al. In Vivo Neuroimaging of Exosomes Using Gold Nanoparticles. ACS Nano. 2017;11:10883-93
46. Yuan H, Fales AM, Vo-Dinh T. TAT peptide-functionalized gold nanostars: enhanced intracellular delivery and efficient NIR photothermal therapy using ultralow irradiance. J. Am. Chem. Soc. 2012;134:11358-61
47. Chithrani BD, Chan WC. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano letters. 2007;7:1542-50
48. Chithrani BD, Ghazani AA, Chan WC. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano lett. 2006;6:662-8
49. Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014;9(Suppl 1):51-63
50. Yuan H, Gomez JA, Chien JS. et al. Tracking mesenchymal stromal cells using an ultra-bright TAT-functionalized plasmonic-active nanoplatform. J. Biophotonics. 2016;9:406-13
51. Kaitsuka T, Tomizawa K. Cell-Penetrating Peptide as a Means of Directing the Differentiation of Induced-Pluripotent Stem Cells. Int. J. Mol. Sci. 2015;16:26667-76
52. Liu Y, Mei L, Xu C. et al. Dual Receptor Recognizing Cell Penetrating Peptide for Selective Targeting, Efficient Intratumoral Diffusion and Synthesized Anti-Glioma Therapy. Theranostics. 2016;6:177-91
53. Lei Y, Tang H, Zhong H. et al. Labeling of hematopoietic stem cells by Tat peptide conjugated quantum dots for cell tracking in mouse body. J. Nanosci. Nanotechnol. 2012;12:6880-6
54. Wu SQ, Chi CW, Yang CX. et al. Penetrating Peptide-Bioconjugated Persistent Nanophosphors for Long-Term Tracking of Adipose-Derived Stem Cells with Superior Signal-to-Noise Ratio. Anal. Chem. 2016;88:4114-21
55. Hansen L, Hansen AB, Mathiasen AB. et al. Ultrastructural characterization of mesenchymal stromal cells labeled with ultrasmall superparamagnetic iron-oxide nanoparticles for clinical tracking studies. Scand. J. Clin. Lab. Invest. 2014;74:437-46
56. Xing X, Zhang Z, Zhong L. et al. Differentiation of human umbilical cord mesenchymal stem cells into steroidogenic cells in vitro. Exp. Ther. Med. 2016;12:3527-34
57. Croitoru-Lamoury J, Lamoury FM, Zaunders JJ. et al. Human mesenchymal stem cells constitutively express chemokines and chemokine receptors that can be upregulated by cytokines, IFN-beta, and Copaxone. J. Interferon Cytokine Res. 2007;27:53-64
58. Reinhard BM, Siu M, Agarwal H. et al. Calibration of dynamic molecular rulers based on plasmon coupling between gold nanoparticles. Nano lett. 2005;5:2246-52
59. Park YI, Im H, Weissleder R. et al. Nanostar Clustering Improves the Sensitivity of Plasmonic Assays. Bioconjugate Chem. 2015;26:1470-4
60. Pascucci L, Cocce V, Bonomi A. et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J. Controlled Release. 2014;192:262-70
61. El-Sayed MA, Shabaka AA, El-Shabrawy OA. et al. Tissue distribution and efficacy of gold nanorods coupled with laser induced photoplasmonic therapy in ehrlich carcinoma solid tumor model. PLoS One. 2013;8:e76207
62. Mooney R, Weng Y, Garcia E. et al. Conjugation of pH-responsive nanoparticles to neural stem cells improves intratumoral therapy. J Control Release. 2014;191:82-9
63. Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nat. Rev. Cancer. 2014;14:683-91
64. Ruan J, Song H, Li C. et al. DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells. Theranostics. 2012;2:618-28
65. Kucia M, Reca R, Miekus K. et al. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem cells. 2005;23:879-94
Author contact
![]() Corresponding authors: wenxiaofei2000com; yuchengedu.cn
Corresponding authors: wenxiaofei2000com; yuchengedu.cn